NC 2023 | 通过遗传编码方法发现可与白蛋白结合并在体内表现出延长循环时间的全氟芳基大环分子
今天介绍的是发表在《Nature Communications》的研究,围绕如何通过遗传编码策略设计具有长循环特性的肽类分子展开。肽类药物因其能够作用于传统“小分子难以靶向”的蛋白相互作用界面而备受关注,但其体内半衰期短、易被快速清除的问题始终限制了应用范围。尽管脂化修饰或与抗体融合等方法可以延长循环时间,这些策略往往会显著增加分子尺寸或影响结构可控性。
该研究提出了一种不同思路,通过全氟芳基化学修饰构建大环肽遗传编码文库,并从中筛选出能够结合血清白蛋白的最小化结构单元。通过结构优化与系统验证,获得了一个仅由7个氨基酸组成的紧凑大环分子,不仅具有微摩尔级亲和力,还能在体内表现出与经典白蛋白结合肽相当的循环时间。进一步将其与功能肽偶联后,显著延长了后者的体内滞留时间。
该工作展示了通过“结构约束+序列筛选”协同设计小分子大环的可行性,为开发兼具小尺寸与长半衰期的新型肽类药物提供了重要思路。

获取详情及资源:
0 摘要
基于肽的治疗分子近年来被视为具有潜力的治疗形式,但其普遍存在的缺陷是在体内循环半衰期较短。该研究从经全氟芳基-半胱氨酸SNAr化学修饰的遗传编码肽库中,筛选获得了能够结合白蛋白的大环肽,其中修饰基团采用十氟二苯砜(decafluoro-diphenylsulfone,DFS)。通过对白蛋白结合能力的测试,鉴定出序列SICRFFC作为核心候选分子。
进一步将DFS替换为等排体五氟苯硫醚(pentafluorophenyl sulfide,PFS),得到的PFS-SICRFFCGG对人血清白蛋白表现出
结果表明,PFS-SICRFFC是目前已知能够与人血清白蛋白结合且具有显著体内循环半衰期的最小肽类大环结构之一,为后续开发具有延长体内半衰期的紧凑型大环分子提供了重要起点。
1 引言
目前全球市场上已有约80种肽类药物,另有超过150种处于临床开发阶段,约400–600种处于临床前研究阶段。与传统小分子药物相比,肽具有更大的表面积,能够作用于蛋白–蛋白、蛋白–糖类以及蛋白–DNA等相互作用中常见的扩展结合界面,这些靶点通常被视为“不可成药靶点”,难以通过小分子实现有效调控,但可以被肽、蛋白或抗体类药物所覆盖。在这三类分子中,肽的分子量最小,通常为2–10 kDa,而完整抗体约为150 kDa,并因此呈现出独特的药代动力学特征。例如,相较于全长抗体,肽和小蛋白在肿瘤及其他缺乏血管的组织中的分布更为均匀,这一优势已在多个临床候选分子中得到体现。
然而,与抗体通过与新生儿Fc受体(FcRn)结合而在体内维持1–3周循环不同,肽类药物通常在数分钟至数小时内通过肾脏迅速清除。这种快速清除在成像、放射性核素递送或短效激素应用中具有优势,但对于更广泛的治疗用途而言,往往需要将其循环时间从分钟级调控至小时级。天然具有较长循环时间的肽较为罕见,例如来源于毒液的39肽exendin-4在人体中的半衰期可达5–7小时,并进一步发展为治疗2型糖尿病的药物exenatide。此后,一系列衍生物通过进一步优化半衰期和药代性质得到开发。总体来看,大多数肽和小蛋白仍需通过修饰来延长循环时间,这些策略主要包括通过PEG或聚甘油等大分子增加尺寸、通过寡聚化调控分子大小、与长循环血清蛋白共价连接,以及引入可与血清蛋白非共价结合的结构单元等。
其中,与血清白蛋白的相互作用尤为关键。白蛋白是血浆中最丰富的蛋白,浓度约为600 μM,在人体中的半衰期约为19天,其长循环特性同样依赖于与FcRn的相互作用,从而避免溶酶体降解并实现循环再利用。白蛋白还能够结合脂肪酸及多种疏水性小分子,从而显著改善药物的药代性质。基于这一特点,通过脂化修饰促进肽与白蛋白结合已成为延长半衰期的成功策略之一,并催生了多种已获批准的药物。然而,除脂类修饰、小分子配体或抗体等形式外,开发分子量更小的白蛋白结合肽仍具有重要意义。
**已有研究报道了一些能够结合人血清白蛋白的肽,例如31肽DX-236、21肽SA-21以及经荧光素和脂肪酸修饰的短肽,这些分子已被用于延长不同配体或蛋白的体内循环时间。**但总体而言,短序列白蛋白结合肽仍然稀缺。为此,该研究采用经化学修饰的大环肽噬菌体展示库,设计了SXCXₙC(n=3–5)的短肽序列,并通过十氟二苯砜(DFS)进行修饰,旨在构建尽可能短的白蛋白结合骨架。研究最初假设全氟芳香结构可作为类似脂肪酸的药效团参与结合,但后续核磁共振及药代研究表明,其主要作用在于约束大环肽构象,使其形成有利于白蛋白结合的结构。该结构单元本身并不能赋予随机肽结合能力,但其构型变化会显著削弱白蛋白结合能力及体内循环性能。
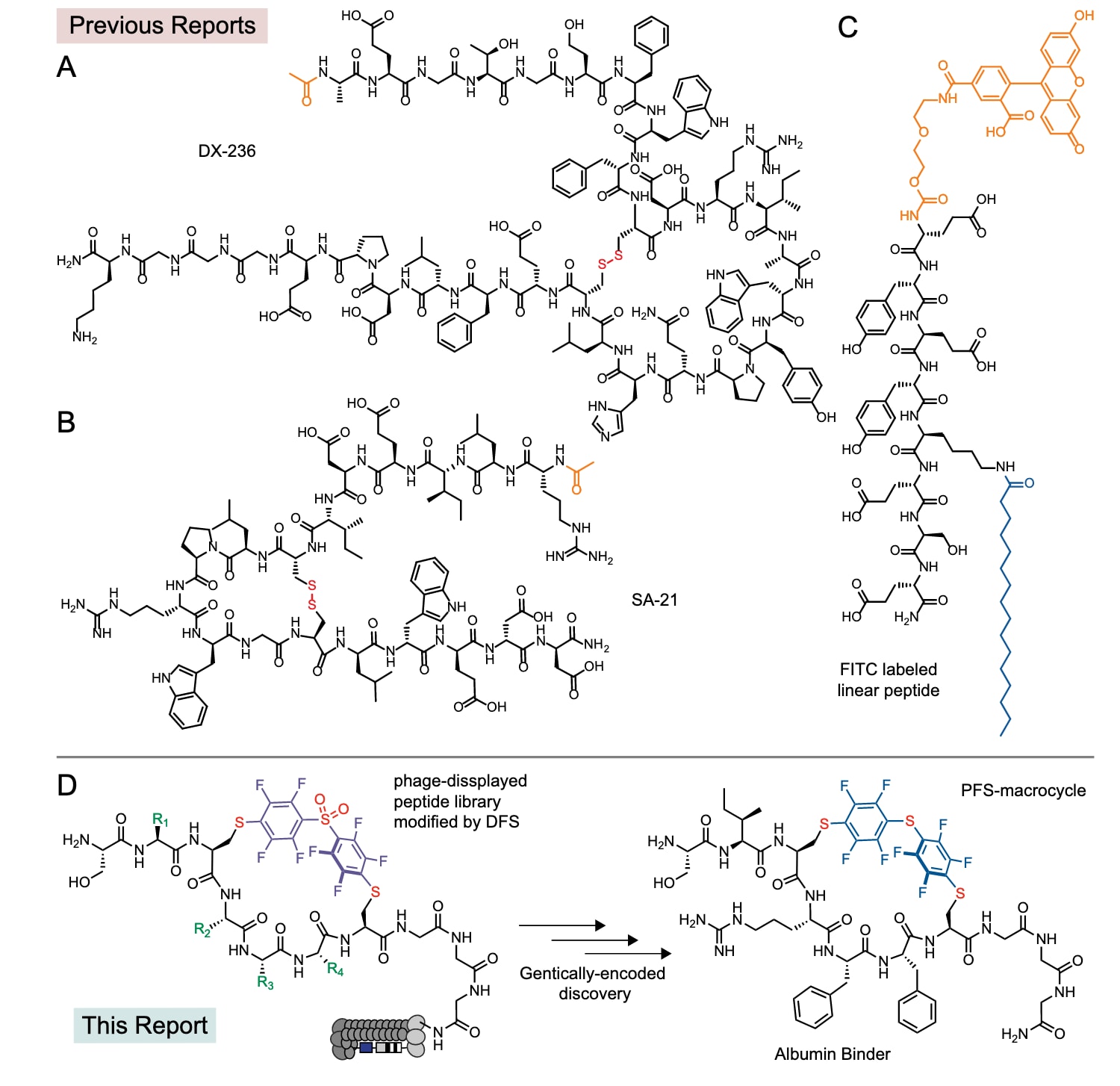
图1|白蛋白结合肽。 已有研究报道了多种白蛋白结合分子:A为大环肽DX-236,B为大环肽SA-21,C为线性肽FITC-EYEYKpalmESE-NH₂。D展示了该研究所采用的方法,即利用经化学修饰的噬菌体展示肽库,用于筛选小尺寸的白蛋白结合大环分子。
2 结果
2.1 白蛋白结合分子的筛选
研究设计并开展了三轮不同策略的筛选实验,分别采用不同的文库结构与筛选方式,以系统发掘能够结合人血清白蛋白的肽大环分子。在第一轮筛选中,按照既有方法对结构为SXCX₄–₅C的噬菌体展示文库进行十氟二苯砜(DFS)修饰,结果表明约85%的文库成功转化为八氟二苯砜交联的大环结构(OFS-SXCX₄–₅C)。随后以固定在96孔聚苯乙烯板表面的白蛋白作为诱饵进行三轮筛选,同时以Protein A作为负对照,以区分特异性白蛋白结合序列与非特异性多蛋白结合序列。第三轮筛选中,OFS修饰文库的回收量相比未修饰文库降低约17倍,说明OFS结构对筛选具有重要影响,但整体富集程度较为有限。尽管进行了Protein A去除步骤,最终富集的序列仍同时结合白蛋白与Protein A,提示特异性不足。
为提高筛选严格性,第二轮筛选调整了实验条件,在不同轮次中交替采用固相板固定的白蛋白与链霉亲和素磁珠固定的生物素化白蛋白。该策略显著提升了筛选效果,在第三轮中回收量较前两轮提高约200倍,而未修饰文库几乎无明显富集。同时,第三轮筛选获得的OFS大环肽在Protein A、ConA及酪蛋白上的结合分别降低2倍、14倍和300倍,表明筛选得到的序列具有较高的白蛋白特异性,且其结合依赖于OFS结构。进一步通过NGS数据进行差异富集分析,识别出多类显著富集的序列家族。第一轮筛选得到的共识序列包括STCHDITC、STCHYIGC和STCHANC,第二轮筛选则得到STCHTIYC。尽管原始文库设计为SXCXₙC(n=4或5),但分析中仍观察到少量SXCX₃C短序列的富集,提示较小环结构可能更有利于结合。
基于这一观察,第三轮筛选专门构建并使用了仅包含SXCX₃C结构的文库,并同样进行DFS修饰,转化率约为88%。该文库规模较小(约16万种变体),能够通过NGS实现全覆盖并进行定量分析。为进一步提高筛选精度,实验采用溶液态白蛋白作为诱饵,避免多价展示效应及固相固定可能带来的构象变化。同时,在筛选体系中加入未标记乳蛋白及His标签融合蛋白,以模拟复杂血清环境。通过链霉亲和素或Ni-NTA磁珠分别捕获白蛋白和对照蛋白,并设置ConA作为对照筛选。最终通过PCR扩增及NGS分析,对不同条件下的富集情况进行比较。
差异富集分析筛选出85条显著富集的候选序列,并通过氨基酸聚类发现以FF为代表的多种结构特征。综合分析后,确定SICRFFC、SFCPMFC和SLCKREC为主要候选序列,并选取STCQGEC作为负对照用于后续合成与验证。总体来看,三种筛选策略获得了不同的结合序列模式,这种差异与蛋白固定方式、去除策略及扩增条件的变化密切相关。随后通过合成这些候选分子,并结合多种生化实验,对其白蛋白结合能力进行了系统评估。
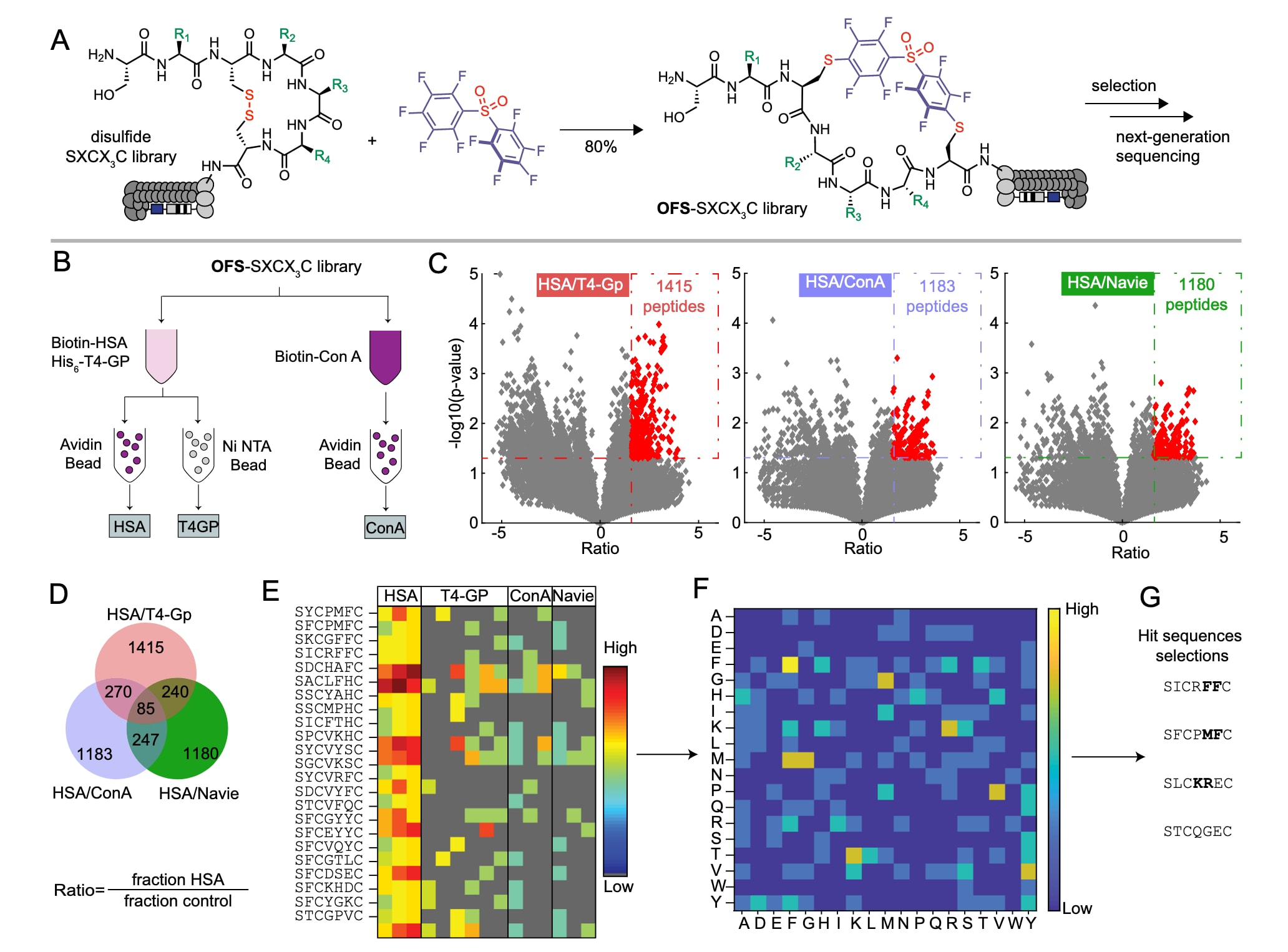
图2|基于化学修饰噬菌体文库的筛选过程。 A展示了将噬菌体展示的SXCX₃C二硫键文库通过DFS修饰,转化为OFS-SXCX₃C大环文库的过程。B描述了筛选策略:在含有未标记乳蛋白的溶液体系中,将OFS-SXCX₃C文库与生物素化人血清白蛋白(HSA)以及带His标签的T4-GP蛋白共同孵育,随后分别通过亲和素磁珠和Ni-NTA磁珠对不同靶标进行捕获;负对照实验中,则以生物素化ConA替代HSA进行筛选并通过亲和素磁珠捕获。C为火山图,D为Venn图,用于展示在HSA筛选中相较于初始文库或针对T4-GP、ConA筛选显著富集的序列。E为差异富集分析中85条候选序列中排名前25的热图展示。F对所有85条候选序列进行了二肽基序分析。G展示了用于后续化学合成及功能验证所选取的代表性序列。
2.2 白蛋白结合分子的验证
在实验过程中观察到,OFS大环肽在碱性条件下会与谷胱甘肽(GSH)等巯基亲核试剂发生非特异性反应。为解决这一问题,将DFS替换为反应性更低的五氟苯硫醚(PFS),得到的PFS大环结构在长达3周内对2-巯基乙醇不发生反应,同时也不会与白蛋白上的游离巯基发生反应。分子动力学模拟表明,OFS与PFS大环在基态下具有相似的构象分布。对于筛选得到的多个全氟芳基交联大环肽(如STCHDITC、STCHYIGC、SICRFFC等),由于其水溶性较差,难以在水溶液中直接评估其与白蛋白的相互作用。因此在其C端引入GGKKK或GGG标签以提高溶解性,并验证这些标签不会显著影响其与白蛋白的结合。
利用全氟芳基修饰所引入的氟信号,可以通过
进一步分析发现,结合能力并非仅由全氟芳香连接基决定,肽序列本身起主导作用。对14c进行丙氨酸扫描显示,R4A或F5A突变会完全破坏与白蛋白的结合,而S1A和I2A突变则分别导致约3倍和10倍的亲和力下降,说明序列中多个残基对结合均有重要贡献。该分子对大鼠和人源白蛋白均表现出类似的结合行为。采用等温滴定量热法尝试测定结合常数时,由于存在多位点、多步骤结合现象,难以获得可靠的定量结果,这一现象与既往研究一致,因此
此外,引入BODIPY荧光基团后,通过荧光偏振(FP)实验进一步验证结合能力。以1 μM肽浓度进行白蛋白滴定,得到PFS-SICRFFCGGK的结合亲和力为
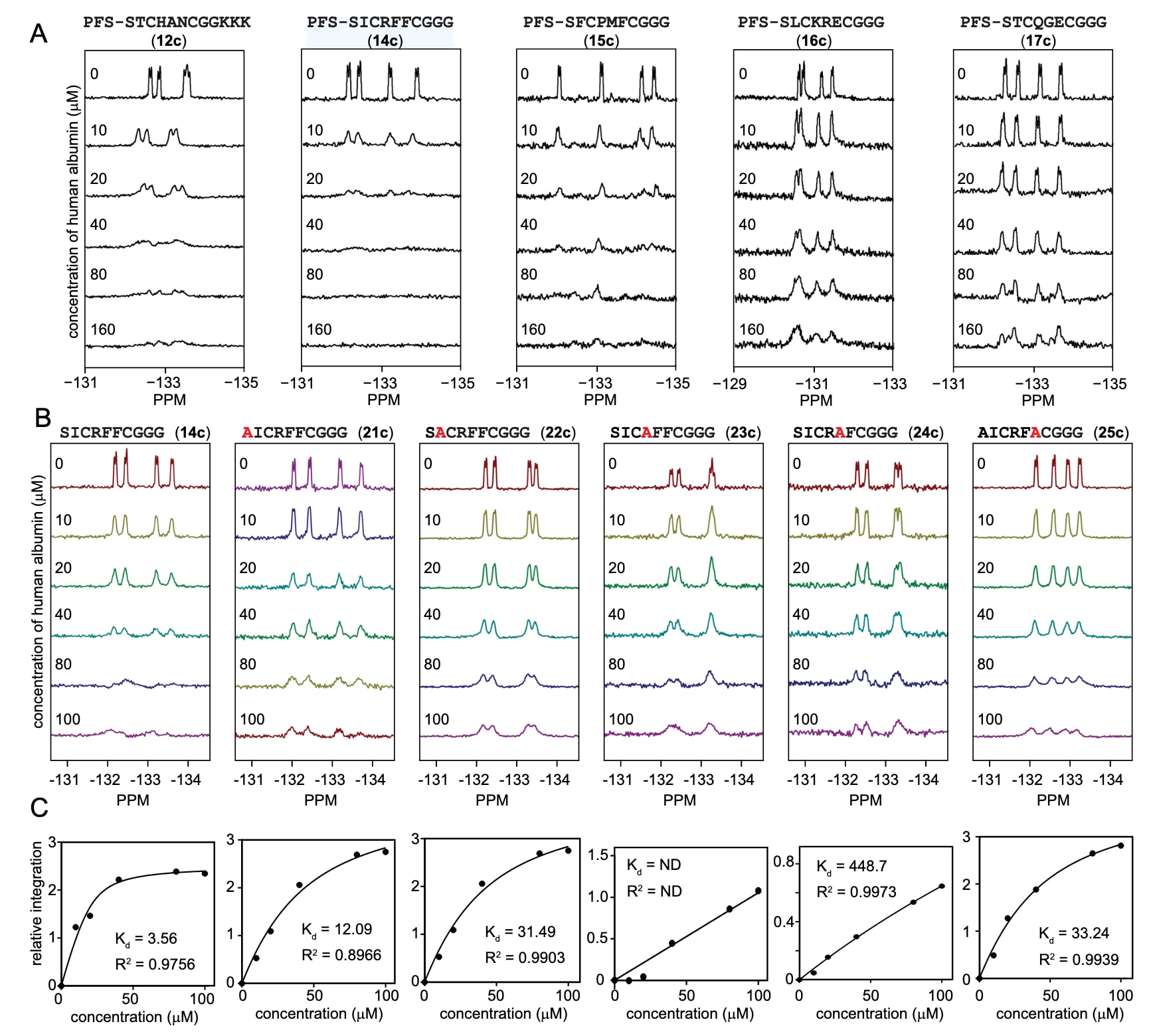
图3|基于
2.3 全氟大环分子的结合位点解析
为确定PFS-SICRFFCGGG(14c)在白蛋白上的结合位置,尝试对其与人血清白蛋白(HSA)进行共晶结构解析,但未获得成功。 因此转而通过竞争实验间接分析其结合位点,考察卡马西平、双氯芬酸和布洛芬是否能够抑制14c与白蛋白的结合。结果显示,这些小分子均未对结合产生明显影响,说明14c的主要结合位点不同于这些药物的经典结合口袋。
进一步通过分子对接计算对白蛋白上的潜在结合位点进行系统评估。已知HSA上存在九个脂肪酸结合位点,其中部分位点也可结合布洛芬或双氯芬酸。对14c在这些位点进行对接分析后发现,位于血红素结合位点附近的“位点1”具有最优结合能力,其平均对接能约为
由于位点1与血红素结合位点重合,理论上可通过血红素竞争实验进行验证,但在
为了进一步验证位点1的合理性,采用分子动力学模拟结合自由能计算方法,对14c在不同位点的结合稳定性进行评估。 在显式水环境下,通过受控分子动力学(SMD)与umbrella sampling计算解离过程的平均力势(PMF),结果显示位点1的结合自由能约为
此外,自由能微扰(FEP)计算得到的突变效应与
这些结果还为后续分子优化提供了重要指导。例如,对接构象及实验结果均表明,14c的N端与C端在结合状态下仍具有一定暴露性,可作为连接药物负载的潜在修饰位点。基于这一认识,后续进一步开展了相关药代动力学研究。
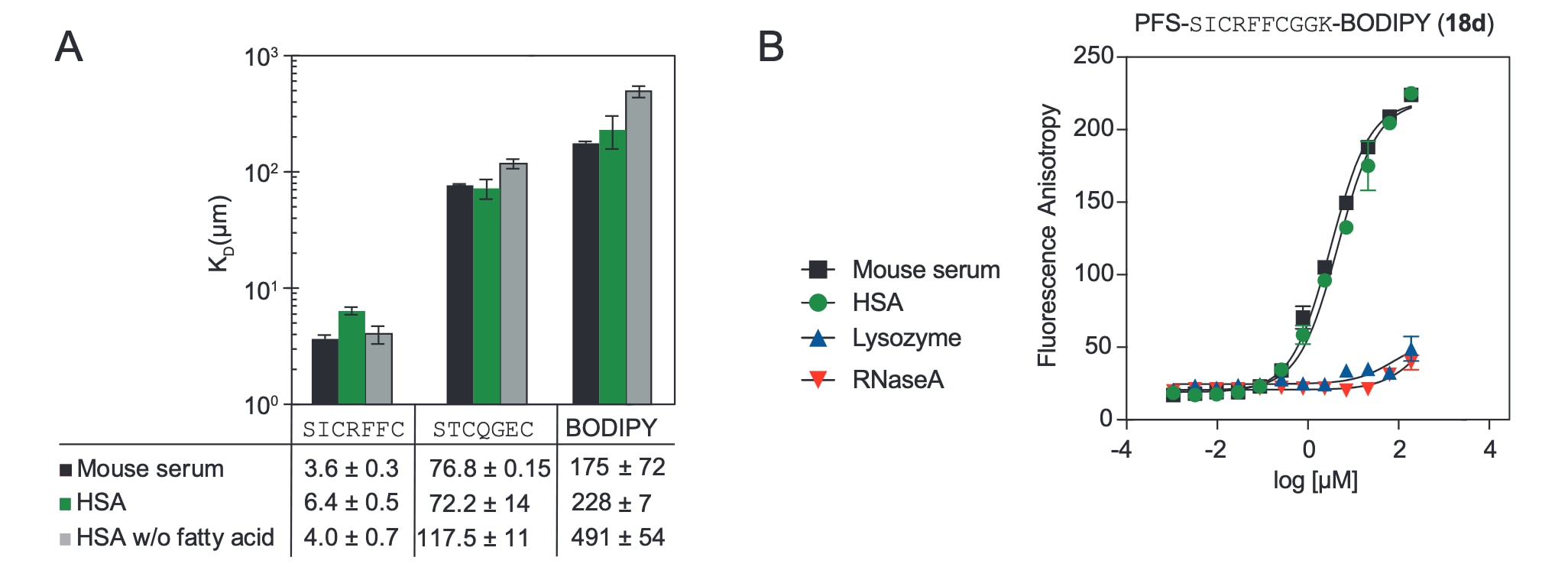
图4|利用荧光偏振测定所选大环分子与白蛋白的结合。 A通过荧光偏振(FP)实验测定了大环分子18d和19d在小鼠血清、人血清白蛋白(HSA)以及去脂肪酸处理的HSA条件下的
2.4 14c衍生物在小鼠体内的循环时间
通过尾静脉注射将14c及其衍生物与SA-21(作为阳性对照)共同给药,并在2分钟和60分钟采集血浆样品进行HPLC/MS分析,从而评估其体内循环表现。结果显示,SA-21在血浆中的浓度仅从
进一步比较不同连接基对体内表现的影响发现,将PFS替换为六氟苯(HFB)或十氟联苯(DFB)虽未完全丧失循环能力,但60分钟时的血浆浓度较14c降低约一个数量级。这些结果表明,14c的体内稳定性依赖于两个关键因素:特定的氨基酸序列以及由PFS连接基赋予的构象约束。
在结构修饰方面,对14c的N端和C端分别进行改造以评估其负载连接位点。N端修饰(如S→A突变或引入PEG基团)会导致60分钟后血浆浓度下降约10倍,同时在NMR中也表现为结合能力下降约3倍,说明N端并不适合作为修饰位点。相比之下,C端修饰表现出更好的兼容性。引入炔丙基甘氨酸或通过点击化学连接PEG链的衍生物(如20c和20h)在60分钟甚至3小时后的血浆浓度均与母体14c相当,而含γ-叠氮赖氨酸的26c则表现较差。此外,带有(Gly)₃连接子的分子通常比(Gly)₂连接子具有更好的循环性能。
在延长时间至3小时的实验中,14c与SA-21的血浆浓度基本一致,而将PFS替换为其他全氟芳香结构或非全氟连接基则会导致显著下降。功能性残基的突变同样显著影响体内表现,例如F6A突变体在60分钟后即无法检测,S1A突变体在3小时后浓度明显降低。与此同时,C端PEG化修饰(如14i和20h)并未影响其长期循环能力,说明(Gly)₃连接子之后的结构变化对结合与循环影响较小。
进一步将14c与apelin-17类似物偶联后发现,其体内表现显著改善。未修饰的apelin-17类似物在注射2分钟后即无法检测,而通过点击化学将其与14c连接得到的化合物(20n)在3小时后的血浆浓度与14c本身相当。这表明,小尺寸的白蛋白结合肽即可显著延长功能肽的体内循环时间,为后续开发高效递送体系提供了重要基础。
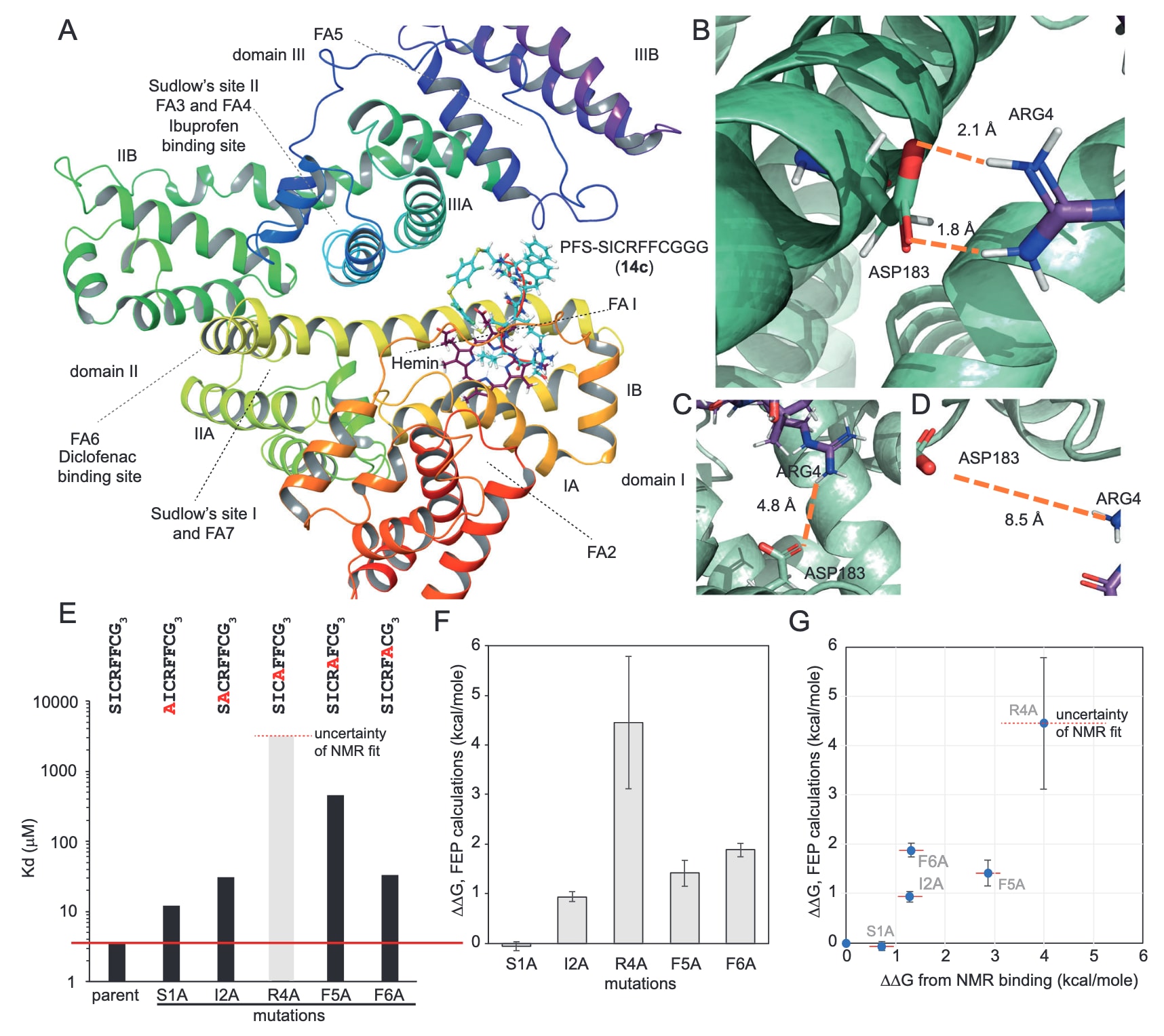
图5|HSA-14c复合物的对接预测。 A显示大环分子14c在白蛋白的多个潜在结合位点中,在靠近血红素结合位点的口袋1具有最低的结合能;同时标注了白蛋白的三个螺旋结构域(DI、DII、DIII)、各子结构域(A和B)以及布洛芬和双氯芬酸的已知结合位点。B中通过受控分子动力学(SMD)与umbrella sampling分析表明,在口袋1中,14c的Arg4与白蛋白的Asp183形成稳定的盐桥;而在其他结合模式中(C、D)未观察到该相互作用。E为通过
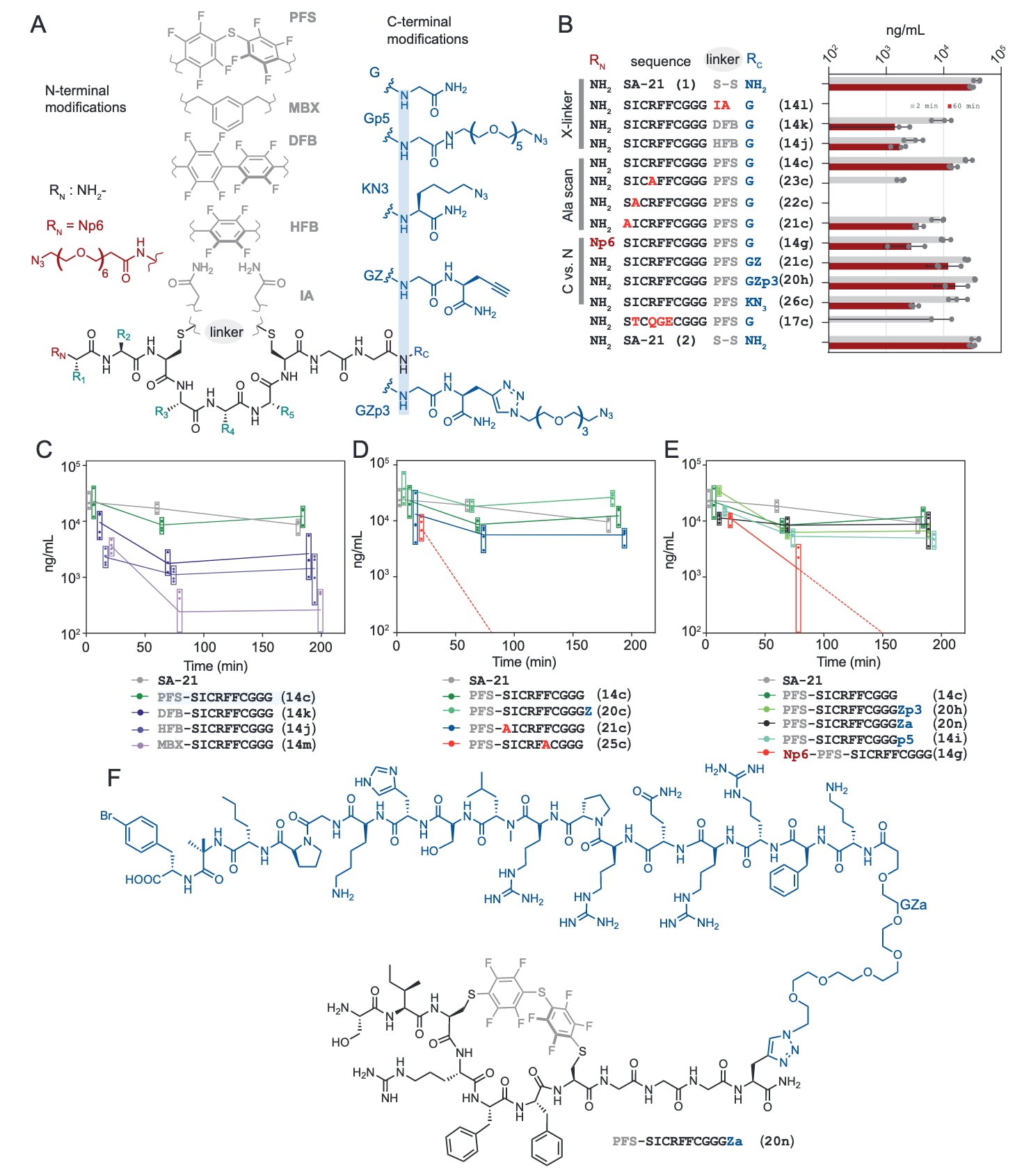
图6|SICRFFCGGG大环分子在小鼠血清中的药代动力学研究。 A展示了核心序列SICRFFC在不同N端和C端延伸以及不同连接基修饰下的结构变化。B中,将包括SICRFFC衍生物、丙氨酸突变体以及SA-21在内的混合物注射入小鼠体内,并在1小时内监测其血浆滞留情况(每种肽n=3)。C分析了PFS-SICRFFC大环在N端和C端引入不同延伸结构用于负载连接时的循环时间表现(每种肽n=4)。D考察了丙氨酸突变以及C端炔丙基甘氨酸修饰对体内滞留时间的影响(每种肽n=4)。E比较了不同连接基对循环时间的影响(每种肽n=4)。F展示了将14c与具有治疗意义的负载分子apelin-17类似物(N3-PEG6-NMe17A2)偶联后的体内表现。柱状图和箱线图均表示平均值±标准差。
3 讨论
通过交联剂(linchpin)对肽及遗传编码肽库进行后期修饰,是赋予其有利性质的常用策略之一。例如,通过SN2反应利用双齿或三齿卤代烷对半胱氨酸进行烷基化,可实现肽的环化、在大环结构中引入非天然片段,以及对噬菌体或mRNA展示文库进行后修饰,从而构建规模达数十亿级的遗传编码文库。另一方面,基于SNAr反应的全氟芳烃介导环化方法可形成芳基硫醚结构,这类方法已被广泛应用,并进一步扩展到生成芳基、烯基及炔基硫醚。与传统的双烷基硫醚相比,芳基及全氟芳基硫醚在氧化稳定性方面更具优势,同时其较低的构象自由度也有助于提升细胞通透性与抗蛋白酶降解能力。
该研究系统探索了基于全氟芳基修饰的大环肽遗传编码文库的筛选策略。此前仅有少数关于SNAr修饰文库的报道,例如利用特定试剂在水相中修饰噬菌体文库,或通过钯催化SNAr反应构建DNA编码文库,但这些方法生成的大环通常不含氟原子。相比之下,全氟芳基交联所引入的氟原子为利用
在筛选过程中还观察到,OFS大环结构对巯基亲核试剂具有一定的温和反应性。近年来,具有弱亲电性的文库及含内建亲电基团的噬菌体文库已成为开发共价或可逆共价抑制剂的重要起点。因此,这种温和反应性在特定情境下可能成为优势,用于筛选能够与蛋白中巯基形成共价键的配体。而在不希望保留该反应性的情况下,可以在筛选完成后将OFS结构替换为近似等排的PFS结构,从而在保持构象与结合能力的同时消除不必要的亲电性。基于此,形成了一种通用策略:首先利用OFS修饰文库进行筛选,然后对命中分子进行PFS替换并验证其结合性能。
白蛋白作为该研究的模型靶标,是噬菌体展示、DNA编码文库及高通量筛选中常用的研究对象。其结构复杂,具有多个结合口袋,能够结合脂肪酸、二羧酸以及多种芳香或杂环小分子,同时也能结合肽和小蛋白,这些特性使其成为延长体内半衰期的重要载体。该研究获得的小尺寸大环肽为白蛋白结合配体提供了新的结构类型。尽管已有小分子可实现极高的白蛋白亲和力,但往往缺乏跨物种结合能力,而该研究中的中等尺寸大环肽则在亲和力与跨物种适用性之间提供了新的平衡。
其中,PFS-SICRFFCGGG(14c)的结合亲和力约为1–4 μM,仅比经典配体SA-21(约0.6 μM)低一个数量级。药代实验还显示,C端(Gly)₃连接子在调控结合能力方面具有重要作用,同时不会显著增加分子尺寸。更为重要的是,14c及其在(Gly)₃之后进一步延伸的衍生物在体内表现出与SA-21相当的循环时间,这一结果突显了其在白蛋白结合应用中的潜力。
由于该类大环肽结构紧凑,既可通过固相合成轻松制备,也可嵌入到其他肽序列中。特别是SICRFFC这一核心基序可以作为固定的N端白蛋白结合模块引入到噬菌体展示文库中,从而构建具有可预测体内半衰期的文库体系。这种紧凑结构还使其能够在筛选早期就被整合到不同的展示平台中,为在动物模型中实现具有可控循环时间的肽及大环分子的从头发现提供了新的可能性。