NC 2025 | protTDA: 蛋白质宇宙的拓扑性质
近年来,深度学习极大提升了蛋白质结构预测能力,AlphaFold2已为超过2亿个蛋白质提供高精度结构模型,使整个“蛋白质宇宙”的结构轮廓逐渐清晰。然而,大规模结构数据如何转化为对蛋白质功能与进化规律的系统理解,仍然是当前结构生物学的重要挑战。该研究提出利用拓扑数据分析作为新的研究视角,通过持久同调方法对AlphaFold2数据库中的全部蛋白质结构进行系统刻画,从整体尺度揭示蛋白质宇宙的组织原则。研究不仅构建了覆盖生命树的蛋白质拓扑图谱,还发现拓扑特征能够精细刻画结构域、预测结合位点,并识别潜在的功能关键区域。此外,拓扑分析还揭示了嗜热蛋白与中温蛋白在结构紧致性上的差异,并能够定位富集疾病相关突变的结构区域。相关工作展示了拓扑方法在理解大规模蛋白质结构数据中的潜力,为研究蛋白质功能、进化及结构稳定性提供了新的理论工具。

获取详情及资源:
0 摘要
深度学习方法已经彻底改变了预测蛋白质结构的能力,使人们得以一窥整个蛋白质宇宙。与此同时,对蛋白质结构如何驱动功能的理解,反而开始落后于结构测定与预测能力的发展。在此背景下,拓扑学这一研究空间结构定性性质的数学分支,被用于作为观察和理解蛋白质宇宙的一个重要视角,从而识别已知蛋白质体系中的基本组织特征。相关研究识别出一系列拓扑决定因素,这些因素能够刻画蛋白质宇宙中的全局特征,例如结构域架构和结合位点。同时,分析还揭示了一些高度特异性的性质,被称为拓扑生成元,它们能够为理解蛋白质结构与功能之间的关系以及进化关系提供更深入的线索。研究提出了一种可在大规模尺度上绘制已知蛋白质宇宙拓扑结构的实用方法,并利用该方法分析突变所带来的结构、功能以及疾病相关后果。该方法不仅揭示并解释了中温生物与嗜热生物蛋白质性质之间的差异,还能够预测蛋白质多态性可能产生的结构与功能影响。在真核生物中,还观察到多细胞生物与单细胞生物之间蛋白质拓扑结构存在显著差异。
1 引言
蛋白质是细胞功能的执行者,也是细胞结构的主要构成单元。蛋白质宇宙指所有生物体中全部蛋白质的集合,其中的每一种蛋白质都具有三维结构;对于部分含有内在无序区域的蛋白质而言,则表现为一组动态变化的三维构象集合。蛋白质科学中的一个基本原则是结构决定功能,因此蛋白质结构的解析在过去七十多年中一直是分子生物学的核心研究内容。随着结构生物学、结构生物信息学以及近年来深度学习方法的发展,大量高精度的结构数据不断积累。AlphaFold2及其不断扩展的一系列相关工具代表了这一领域的重要突破。AlphaFold2是一种用于预测蛋白质结构的深度学习模型,在预测精度和规模上均显著超越以往的方法。目前,AlphaFold2数据库已包含来自生命各个界的超过2.14亿个独特蛋白质结构模型,因而很可能已经覆盖了几乎整个蛋白质宇宙。然而,对如此庞大规模的结构数据进行直接分析在过去几乎是不可能完成的任务。随着序列比对算法计算能力的显著提升,例如MMseqs2的发展,序列信息如今可以作为分析AlphaFold2数据库的重要指导原则。例如,可以通过序列信息对蛋白质结构进行聚类分析,或在序列聚类的基础上对部分AlphaFold2结构进行进一步的结构比对。结构生物信息学与深度学习方法的结合因其重要贡献而获得了2024年诺贝尔化学奖。然而,目前常用的大多数蛋白质结构分析工具在设计之初主要针对规模较小的数据集开发,尽管也存在少数例外,但尚未有工具能够对整个AlphaFold2数据库进行系统性的结构分析。这一现状表明,有必要开发新的方法以揭示蛋白质宇宙中的组织原则。
蛋白质结构可以用拓扑学的概念进行描述。拓扑学为理解蛋白质中二级结构元件(如α螺旋、β链和β折叠)之间的连接方式与空间排列提供了一种强有力的框架。通过对这些二级结构及其相互关系进行映射,可以将复杂的三维结构简化为一种更加抽象的表示方式,从而有助于识别不同生物体或不同蛋白质家族中反复出现的结构基序、空间排列模式以及功能区域。因此,对蛋白质拓扑特征的分析是蛋白质科学的重要基础之一,常用于理解结构与功能之间的关系、推断蛋白质的进化联系,以及设计具有新功能的蛋白质。在数学中,拓扑学关注空间结构的定性特征,例如连通性以及孔洞或空腔的存在。拓扑学认为,如果两个结构能够通过拉伸、弯曲或扭转相互转化,而无需切割或粘合,则它们在拓扑意义上是等价的。拓扑视角的优势在于能够识别那些不依赖于具体空间尺度或时间尺度的关键特征。对于蛋白质而言,如果两个蛋白质通过相似的口袋和相似的相互作用方式结合相同的配体,那么无论它们在大小或整体三级结构上存在何种差异,都可以被视为拓扑上等价的结构。因此,利用数学拓扑方法分析蛋白质结构,有望在复杂的多维数据中发现潜在或隐藏的结构模式。
尽管这种描述看起来较为抽象,但拓扑方法在多个领域已经显示出重要价值。例如在物理学中,Morse理论和Floer同调为量子场论与宇宙学中的基本规律提供了深刻的数学结构。近年来,拓扑数据分析(Topological Data Analysis,TDA)逐渐成为数学拓扑学中的一种重要新方法。TDA的核心思想是利用代数方法分析数据的“形状”。其中最有效的技术之一是持久同调(Persistent Homology,PH)。这种计算方法能够将离散分布的数据点转化为一系列逐渐变化的几何结构,从而识别在不同尺度下持续存在的系统特征。当这一方法应用于空间对象(例如蛋白质结构)时,可以观察随着数据点空间范围逐渐扩大并发生重叠时,整体结构形态如何演化并形成新的模式。在这一过程中,PH记录拓扑特征的产生与消失,并通过“持久性”这一指标来衡量特征存在的时间长度。持续时间越长,说明该特征越可能反映真实的数据结构,而非随机噪声。所有这些特征及其持久性数值共同构成了描述系统结构的重要指标,并已被证明在自然科学与物理系统中的聚类分析、参数推断和模式识别方面具有很高的有效性。
在此基础上,相关研究开发并优化了一种基于持久同调的拓扑数据分析方法,用于系统分析AlphaFold2预测的全部2.14亿个蛋白质结构。通过这一方法,可以从统计层面提取蛋白质宇宙中的组织原则以及拓扑结构与功能之间的关系,并构建一个用于探索AlphaFold2庞大结构资源的“拓扑导览”。这一策略为当前规模最大的蛋白质结构数据集提供了一种系统化的分析框架,使得在前所未有的规模上研究蛋白质结构与功能之间的联系以及蛋白质进化规律成为可能。
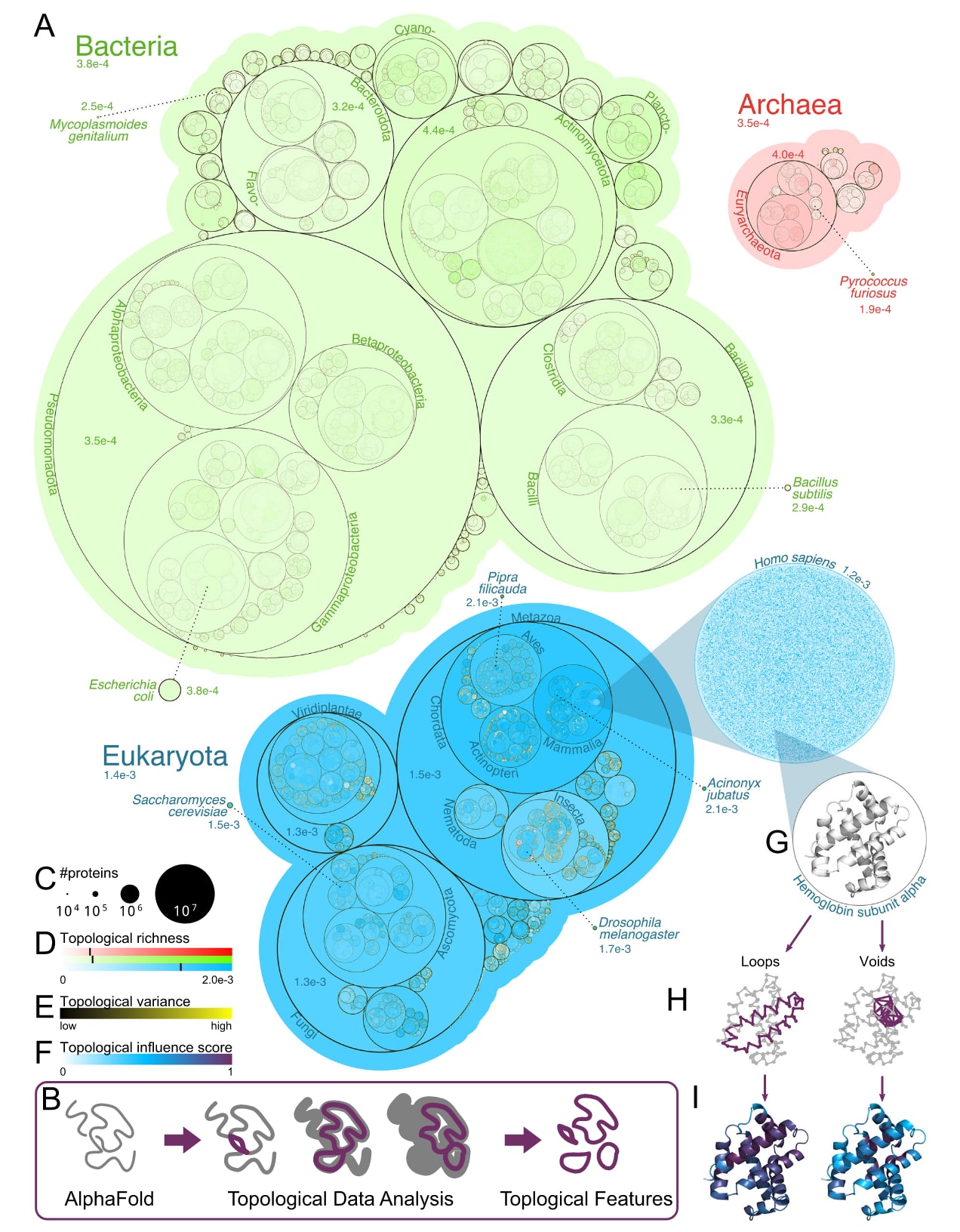
图 1 | 蛋白质宇宙具有丰富的拓扑结构。 图中展示了 2.14 亿个 AlphaFold2 预测的蛋白质结构,它们按物种分类并绘制成生命树(A)。对这些结构的拓扑分析(示意于紫色框内,B)揭示了拓扑特征的复杂性与多样性,以及在进化树上呈现出的显著复杂度,这里采用圆形堆积图加以呈现。每个圆形的面积与该类中蛋白质的数量成正比(C)。圆形的饱和度代表该类蛋白质的平均拓扑丰富度,并在域、界、门以及部分感兴趣的物种中给出数值。平均丰富度通过以类内蛋白质平均尺寸进行归一化来近似计算。颜色尺度的上限设为 95% 分位数,以忽略异常值并突出差异,并以黑色线条标示各域的平均值(D)。圆形的边界颜色反映拓扑方差(E)。放大到人这一类群时,每个蛋白质以点表示,颜色饱和度与其拓扑丰富度成正比。血红蛋白(G)被单独绘制,展示其最持久的一维拓扑特征(左侧,一个环)与二维拓扑特征(右侧,一个空腔)(H),下方则以氨基酸的颜色对应其拓扑影响得分(I),颜色尺度为白—蓝—紫渐变(F)。
2 结果
2.1 蛋白质宇宙拓扑分析流程的建立揭示其丰富的拓扑结构特征
持久同调研究的一个最新进展是能够高效地确定“同调生成元”,并对其进行系统分析。拓扑生成元可以精确定位数据中产生拓扑特征的具体区域或关键结构。在单个蛋白质层面,这些生成元能够揭示形成高阶结构特征的氨基酸相互作用群,例如特定构象,或结状蛋白中的结构缠结。该研究在此基础上扩展了该方法,对AlphaFold2数据库中超过2.14亿个蛋白质结构进行了系统分析。为了能够处理规模前所未有的拓扑生成元数据,开发了一套用于批量计算持久同调并优化内存使用的计算流程。随后对拓扑结果的分析采用了此前提出的方法,并按照图1B和补充图3所示的流程进行。
在该流程中,首先利用蛋白质的α碳原子对结构进行建模,将蛋白质表示为点云结构。点云表示的优势在于能够将复杂的三维结构简化为坐标空间中的一组点,每个残基对应一个
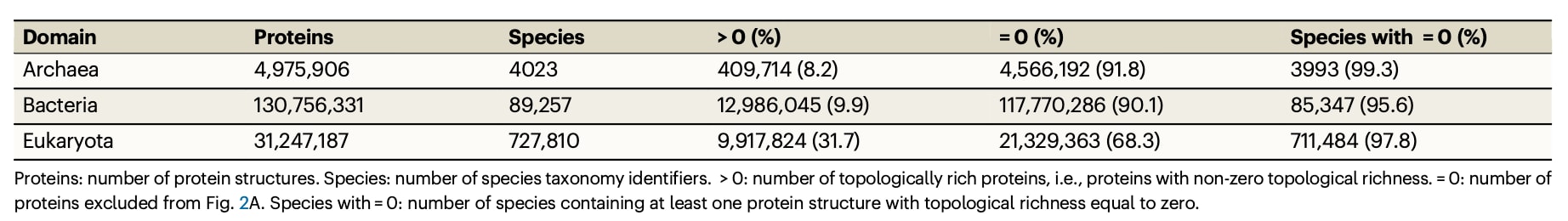
整个计算过程共消耗约10560个CPU小时,并在Oracle Cloud Compute平台上完成。这些计算产生了超过9.85TB的拓扑数据,并构建了目前已知蛋白质宇宙的拓扑图谱,相关数据已经公开发布。为了在最宏观的层面理解这些数据,进一步构建了“生命拓扑树”。该结构以圆形嵌套图进行可视化,每个圆的面积代表对应物种在AlphaFold2数据库中预测的蛋白质结构数量。随后按照属级别对不同物种进行连接与排序,使不同等级区域的面积大致反映结构数量。生命三域——细菌、古菌和真核生物——在该拓扑树中均得到充分表示,并涵盖了蛋白质组规模差异极大的生物。
进一步对每个蛋白质、每个物种以及不同生命域的拓扑丰富度进行了系统测量。拓扑丰富度用于描述蛋白质中独特且持久的拓扑特征数量,并对所有蛋白质求平均后按残基数量进行归一化。结果显示,细菌和古菌蛋白质整体上表现出较低的拓扑丰富度,而真核生物则存在多个拓扑丰富度显著升高的区域,尤其在哺乳动物类群中更为明显。一些物种表现出特别突出的特征,例如猎豹(Acinonyx jubatus)和细尾伞鸟(Pipra filicauda)。相比之下,人类蛋白质在哺乳动物中反而呈现相对较低的拓扑丰富度。虽然这一现象看似出人意料,但类似于人类基因数量相对较少的现象,这可能反映拓扑复杂性只是衡量生物复杂度的一种方式。人类在蛋白质层面的复杂性很可能更多来自多层次调控机制、发育程序、可变剪接以及复杂的蛋白质相互作用网络。
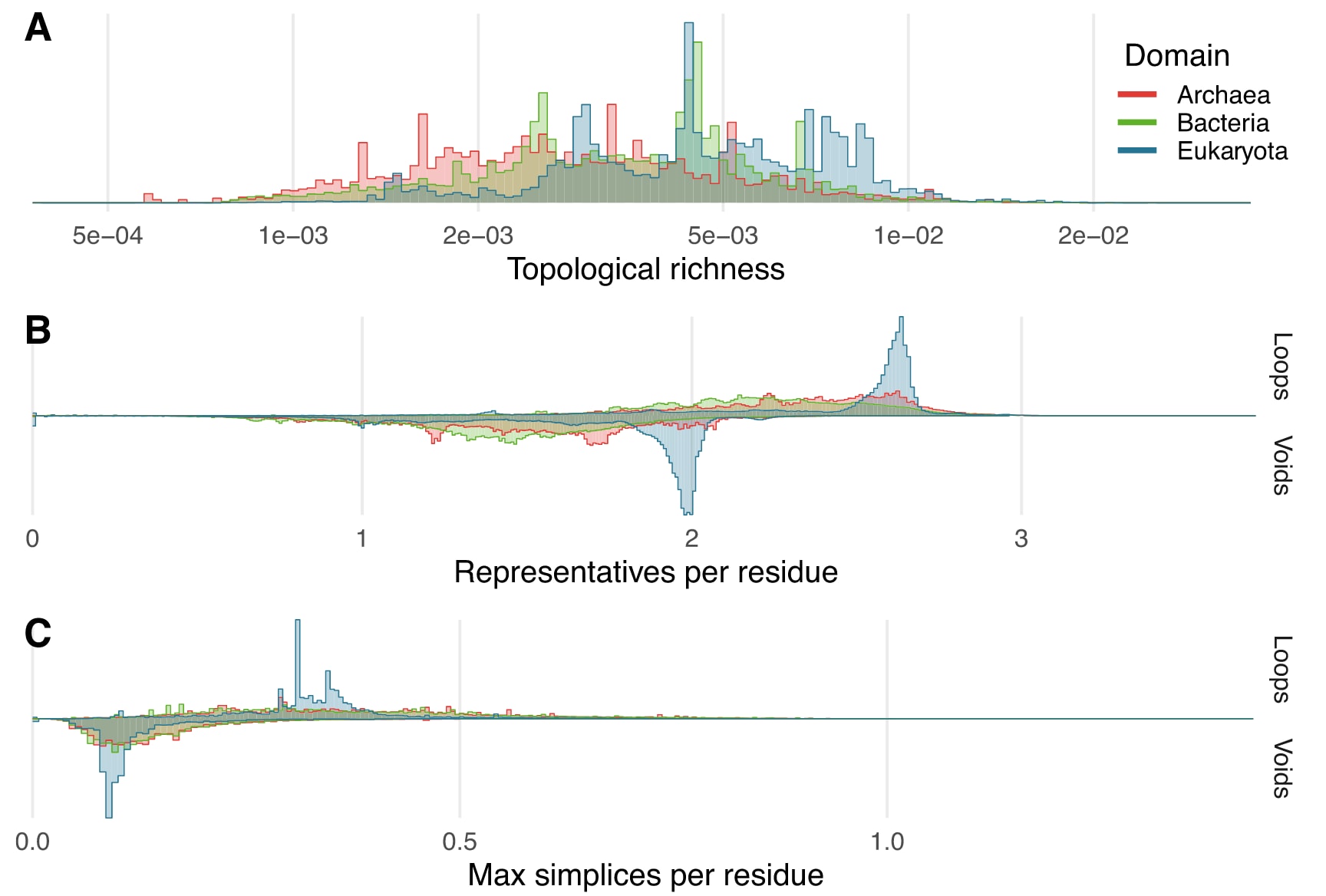
图 2 | 166 979 444 个高质量结构的拓扑特征分布。 对于非零丰富度的蛋白质,来自不同域的高质量蛋白质在拓扑丰富度上呈现出轻微偏移(A)。该图以对数刻度呈现,零值条目的排除数量见表 1。拓扑代表的数量——即“环”或“腔”——经蛋白质长度归一化,用于指示平均残基的拓扑特征归属情况(B)。最大单个环和腔的规模以单纯形数量衡量,并同样经蛋白质长度归一化(C)。就环而言,这大致对应于蛋白质中属于其最大环部分的残基比例。所有子图的分箱频率计算均给予各物种相同权重。每个密度图均为经过缩放的直方图,其总面积为 1。低质量结构已通过平均 pLDDT 大于 70 的阈值过滤。结果显示,真核生物的蛋白质在拓扑倾向上与其他两个生命域不同,通常表现出更高的拓扑复杂性。源数据以文件形式提供,位于 Source_data/source_data_Figure2ABC.tsv。
在数百万蛋白质上系统整理拓扑特征,使得从蛋白质宇宙尺度定量研究蛋白质性质成为可能。整体分析显示,真核生物与细菌和古菌之间存在明显差异。当只关注拓扑丰富度较高的蛋白质时,各生命域的分布略有变化。总体而言,真核蛋白质结构更加复杂,而古菌蛋白质虽然包含较多拓扑特征,但结构复杂度相对较低。由于拓扑丰富度的计算采用较高阈值的环结构统计,大多数蛋白质的丰富度值接近零,因此未出现在密度分布图中。
通过比较单个氨基酸平均参与的环和空腔数量,可以观察到真核蛋白质中存在大量具有较高成员数量的拓扑结构,而细菌和古菌则表现为更均匀且平缓的分布。进一步分析每个蛋白质中最大环和最大空腔的规模发现,真核蛋白质仍然表现出明显差异。最大环的大小通过简单复形数量与残基数量之比进行估计,即表示蛋白质序列中包含在最大环结构中的比例。结果表明,真核蛋白质较少将结构集中在单一的大环或空腔中,而更倾向于形成多个环和空腔,从而表现出更高的拓扑复杂性;相比之下,细菌和古菌的拓扑特征分布则更加均匀。
更为显著的现象是真核生物分布中出现了明显的峰值,而细菌和古菌的分布整体较为平滑。在如此大规模的跨物种数据中,均匀分布本应更为常见,因此这些尖锐峰值显得尤为突出。这一现象可能反映了多细胞生物在实现复杂调控机制时所偏好的特定蛋白质复杂度水平。为进一步分析这一现象,将真核数据划分为两组:一组包含后生动物和陆地植物,即大致代表多细胞生物;另一组包含其余真核生物。结果表明,这些明显峰值主要出现在多细胞生物这一组中。虽然数据集中的潜在偏差,例如某些被大量研究的蛋白质家族,也可能导致类似现象,但由于分析中对不同物种赋予相同权重,因此可以避免研究较多的模式生物对结果产生过度影响。
研究还对拓扑方差进行了绘制,该指标可以视为拓扑特征在进化过程中稳定性的度量。拓扑方差定义为某一圆形区域中一维拓扑特征数量的方差,并按该区域蛋白质数量进行归一化。结果显示,真核生物的拓扑方差整体高于细菌和古菌,在昆虫类群中尤为明显,这表明其拓扑结构特征具有更高的多样性。相反,在细菌中如果不同分类层级的方差始终较低,则可能说明其拓扑复杂度在进化过程中保持相对稳定。
最后,将拓扑影响评分映射到所有蛋白质结构上,从而获得残基层面的拓扑信息。在单个蛋白质中,TIF值反映每个残基在拓扑结构中的重要程度,这有助于识别结构关键区域以及潜在的致病突变位点。在具体实例中,例如人类血红蛋白α亚基的结构分析中,可以识别出最持久的环和空腔结构,并通过TIF值观察它们对整体拓扑结构的影响。
整体而言,该方法为分析蛋白质宇宙的拓扑结构提供了一种强大而灵活的工具。通过对AlphaFold2数据库中接近四分之一十亿个蛋白质结构进行系统分析,揭示了生命树范围内蛋白质拓扑特征的多样性与复杂性。
表 1 | 来自各个域的拓扑丰富蛋白质
2.2 蛋白质宇宙的拓扑分析能够实现更加细致的蛋白质结构解析
近期研究表明,持久同调分析可以与网络理论相结合,从而揭示不同拓扑特征之间更深层次的联系。基于这一思想,对蛋白质宇宙的拓扑图谱进一步进行网络化解释。在该框架中,将氨基酸残基视为节点,而由环(1维拓扑特征)和空腔(2维拓扑特征)定义节点之间的连接关系。连接的强度及其重叠程度会使氨基酸在结构上形成若干聚集单元,这些单元被称为“拓扑簇”。这种方法能够捕捉蛋白质宇宙中的整体结构特征,并识别传统蛋白质结构分析方法难以发现的结构性质。
研究发现,1维拓扑簇(即由环结构定义的簇)与蛋白质结构域密切相关,而结构域通常被认为是蛋白质中相对独立的折叠单元,这一点可以通过CATH蛋白质结构分类数据库进行验证。以一个蛋白激酶(UniProt编号Q4DF08)为例,对其CATH结构域与拓扑簇之间的关系进行分析。结果显示,在这一蛋白以及许多其他蛋白中,1维拓扑簇能够很好地反映CATH结构域的基本特征,但同时提供更高分辨率的信息。例如,一个CATH结构域往往可以被进一步划分为多个拓扑簇,说明拓扑分析能够在传统结构域划分的基础上提供更加精细的结构划分。
为了定量评估拓扑簇与CATH结构域之间的一致性,引入同质性评分进行比较。当拓扑簇能够完全对应结构域划分时评分为1,而当两者完全无关时评分为0。研究分析了38171个AlphaFold2预测的蛋白质结构,这些结构代表不同蛋白质家族、结构域和生物物种,并且均为非冗余且置信度较高的预测结果,同时至少包含两个已识别的CATH结构域。结果表明,大多数拓扑簇都位于单一结构域内部,因此拓扑分析通常是在结构域划分的基础上进一步细化结构特征,揭示许多结构域实际上由多个不同的拓扑结构组成。这一发现对于蛋白质进化研究和蛋白质工程具有潜在重要意义,因为这些领域通常将结构域作为分析的基本单位。整体结果表明,数学拓扑分析与CATH等数据库中记录的蛋白质结构域总体一致,同时在许多情况下能够提供更加精细的结构特征。由于CATH结构域与蛋白质折叠过程密切相关,因此可以进一步推测,1维拓扑簇可能对应单独的折叠单元。
除去0维拓扑中简单的连通成分统计外,最基本的拓扑特征通常来自1维结构(即环)。这种特征能够有效捕捉结构形状的定性空间信息。在蛋白质体系中,以往研究表明环结构能够反映复杂的几何子结构,例如结构缠结以及其他非平凡空间特征。当前研究的结果与这一观点一致:1维环结构在CATH结构域内部往往高度交织,而跨越不同结构域的环结构在拓扑聚类过程中通常会被分离。尽管实验上获取蛋白质折叠中间体十分困难,但已有研究显示,脱辅基肌红蛋白在微秒到毫秒尺度上可以形成部分折叠结构。在这一案例中,1维拓扑分析成功识别出初始折叠核心区域。这一结果初步表明拓扑方法能够为蛋白质折叠研究提供补充视角。然而需要注意的是,AlphaFold结构预测并不能提供反映蛋白质折叠动力学所需的构象集合信息。
与能够揭示局部结构单元的1维拓扑特征不同,2维拓扑特征(空腔)可能与配体结合位点相关。为了验证这一假设,分析了空腔拓扑簇与结合位点之间的距离关系。结合位点来自Mechanism and Catalytic Site Atlas(M-CSA)数据库。研究共分析了866个AlphaFold2预测的蛋白质结构(其中862个为高置信度预测),这些结构涵盖多种酶家族以及其他能够结合配体的蛋白质。具体方法是将RCSB蛋白质结构数据库中1033个具有M-CSA注释位点的实验结构映射到UniProt序列,然后选择其中对应的高置信度AlphaFold2预测结构。通过计算空腔边界与结合位点之间的残基距离发现,大约70%的结合位点位于空腔边界处或仅相隔一个氨基酸残基。
这一结果在结构层面具有合理性,因为配体结合位点通常位于结构可接近且具有一定柔性的区域,而拓扑分析能够在2.14亿个预测蛋白质结构中系统识别这类区域。因此,对空腔结构的分析有望帮助发现潜在或隐藏的结合位点。
尽管AlphaFold2在结构预测方面具有极高精度,但在某些蛋白质体系中仍可能无法完全捕捉结构复杂性。持久同调方法的重要优势之一在于其鲁棒性:即使输入数据存在一定程度的扰动,其拓扑特征仍保持相似。因此,可以合理认为拓扑分析不会过度依赖局部三维构象的细微误差。为验证这一点,将RCSB蛋白质结构数据库中的实验结构与对应的AlphaFold2预测结构进行比较,并通过1维和2维拓扑影响评分进行评估。结果显示,两者在逐残基层面的TIF值具有高度相关性,这表明拓扑分析结果能够在实验结构与预测结构之间良好迁移。
整体而言,这些结果展示了拓扑方法在识别蛋白质结构组织特征方面的价值,并证明其能够为蛋白质结构分析提供新的视角与工具。
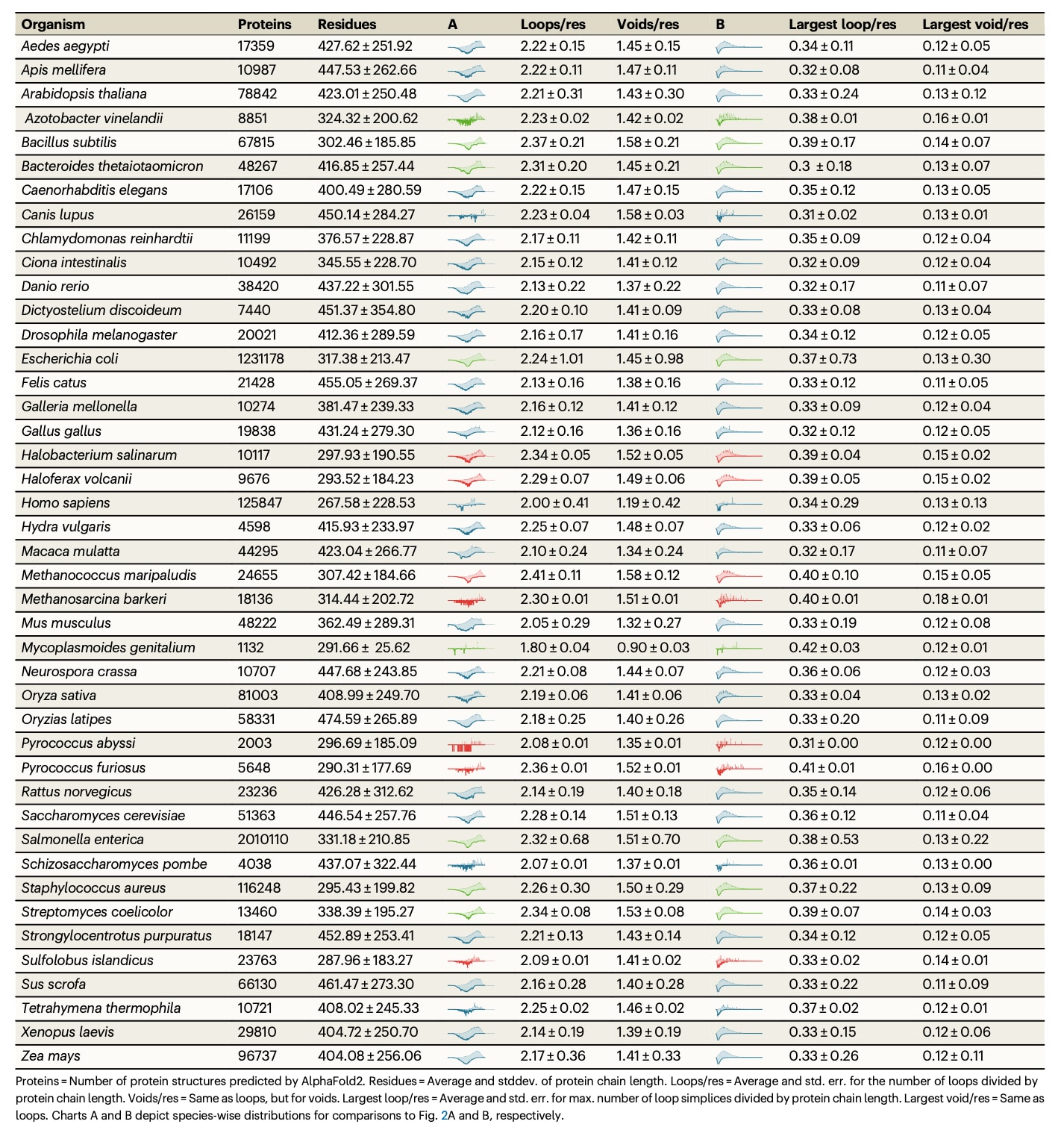
表 2 | 模式生物的分布
2.3 嗜热蛋白与中温蛋白的拓扑比较
嗜热蛋白如何在保持功能的同时实现结构稳定性,一直是蛋白质科学、结构生物学和进化生物学中的重要问题。已有研究提出多种可能的决定因素,例如疏水性差异、二级结构比例、离子对、氢键以及蛋白质内部空腔的数量和大小等。然而,由于统计样本不足以及潜在混杂因素难以严格控制,这些因素的作用仍存在较大争议。随着AlphaFold2产生的大规模结构数据以及拓扑分析方法的鲁棒性,相关研究提出可以通过拓扑特征检测嗜热蛋白与中温蛋白之间的结构差异,即使这些蛋白质在结构上高度相似。这些差异有望揭示嗜热蛋白在高温环境中维持结构与功能的机制。
寻找这种差异并不容易,因为在不同生物体中,同一类酶往往具有几乎完全重叠的三维结构。例如葡萄糖-6-磷酸脱氢酶在大肠杆菌(中温生物)和M. thermoacetica(嗜热生物)中的结构几乎完全一致。为系统比较两类蛋白质,研究选择了10类在生物技术中具有重要意义的酶(根据酶学委员会EC编号分类),这些酶来自30种嗜热生物和8种中温生物,共计1656个高置信度AlphaFold2结构预测。分析重点放在二维拓扑特征,即空腔结构上,因为空腔的紧密程度可能与蛋白质在高温条件下的稳定性相关。此外,为减少蛋白质序列长度、底物性质和功能差异等因素带来的干扰,仅比较序列长度相同的同源蛋白。
结果显示,嗜热生物蛋白质中的空腔通常比中温生物更小、更紧凑。通过在排除持久性小于1Å的噪声后进行单侧Mann–Whitney U检验,发现这种差异具有显著统计意义。进一步通过在每个EC类别中随机抽取等量样本进行对照分析,结果仍然显示显著差异。这说明嗜热蛋白的拓扑空腔确实整体更为紧密。
随后进一步分析这种差异是否可能由空腔周围氨基酸体积的变化所补偿。通过比较二维同调中基于TIF计算得到的氨基酸贡献分布,可以间接反映空腔附近氨基酸的丰度。结果显示,虽然中温和嗜热蛋白在不同氨基酸上的分布形状存在显著差异,但TIF值与氨基酸体积之间仅表现出较弱相关性,Pearson相关系数约为0.155,这表明较大氨基酸在空腔附近略微更常见,但这种趋势较弱且在两类蛋白质中基本一致。进一步的相关性检验均得到极显著的统计结果,但回归分析显示两组回归斜率的95%置信区间高度重叠,因此无法证明氨基酸体积能够补偿空腔体积差异。尽管如此,嗜热蛋白的氨基酸体积中位数略高于中温蛋白(140对138.4),这一差异在统计上显著,同时回归斜率也略高,说明氨基酸体积可能在一定程度上进一步增强蛋白质结构的紧密性。
综合这些结果可以推测,嗜热蛋白与中温蛋白之间的拓扑差异可能反映了不同环境中热力学压力的影响。在高温环境下,如果结合口袋内部存在较大的空腔,既可能降低结合特异性,也可能削弱结构稳定性。因此,较小且紧凑的空腔结构可能有助于维持嗜热蛋白在高温条件下的稳定性和功能。
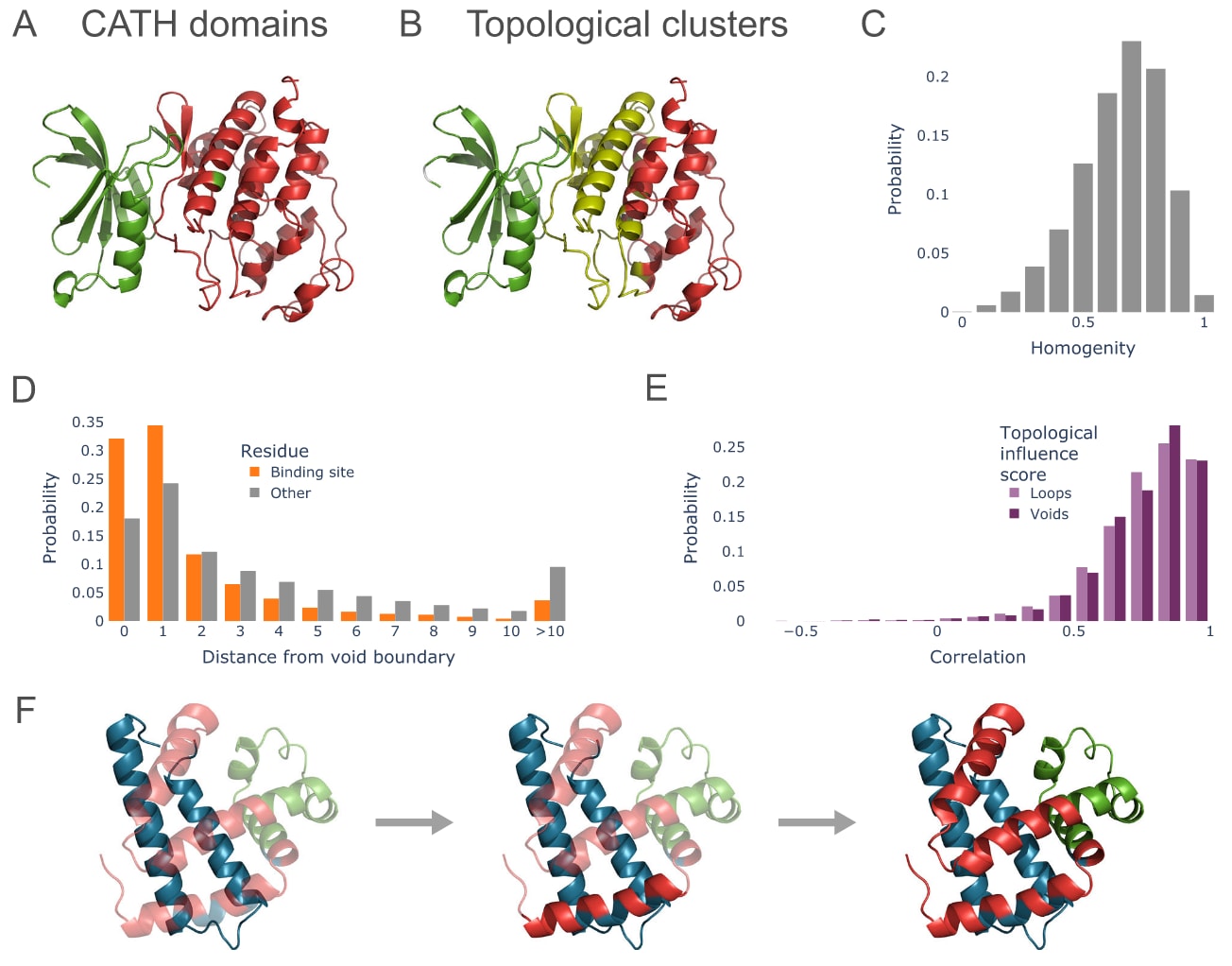
图 3 | 拓扑结构为预测蛋白质结构提供了组织原则。 一维拓扑簇往往能够对蛋白质结构域进行细化划分。例如,蛋白质激酶按照其两个 CATH 结构域着色(A),并可对照其拓扑簇着色结果(B)。聚类同质性可用于检验一个拓扑簇是否仅包含归属于单一结构域的残基,数值 1 表示完美的子分区,0 则表示簇内的标签完全一致。柱状图显示了 38 171 个具有已识别 CATH 结构域的非冗余 AlphaFold2 预测模型中同质性分数的分布情况(C)。二维拓扑簇的边界点在机制与催化位点图谱(M-CSA)数据集的结合位点处富集。柱状图展示了簇边界与结合位点残基或其他残基之间的距离分布(以残基数计)(D)。拓扑分析对小扰动具有稳健性。柱状图显示了 AlphaFold2 预测结构与实验解析结构之间拓扑影响分数的相关系数分布。较高的相关系数表明拓扑特征倾向于对应相同的残基,并且关注的区域一致(E)。马阿朴肌红蛋白(UniProt 登录号 P68082)的拓扑簇通过颜色区分(F)。基于实验证据的折叠事件以透明度标记,完全不透明的区域表示已形成结构。源数据分别提供于 Source_data/source_data_Figure3C-S8.csv、Source_data/source_data_Figure3D.csv 和 Source_data/source_data_Figure3E.csv。
2.4 拓扑分析识别富集疾病相关突变的蛋白质区域
由于蛋白质功能依赖于结构与序列,因此进一步研究拓扑分析是否能够识别富含致病突变的结构区域。为验证这一点,使用一个包含致病突变和中性突变的数据集,该数据集收录了数百种野生型和突变蛋白质的实验结构,并曾被用于研究突变对结构稳定性的影响。分析同样仅限于高置信度AlphaFold2预测结构。
在每个蛋白质中,需要识别那些在结构上具有重要作用的残基,因为这些位置更可能在发生突变时导致结构破坏,从而产生疾病相关多态性。TIF值可以衡量每个残基在拓扑结构中的重要性,因此一个关键问题是较高的一维或二维TIF值是否意味着更大的结构稳定性影响。
整体结果显示,与非致病突变或无结构作用的多态位点相比,导致结构改变、导致疾病或同时导致结构改变和疾病的突变更可能出现在拓扑生成元附近。以人类ACE2和血红蛋白HBB为例,将蛋白质结构按照二维TIF值进行着色,可以观察到结构重要区域与致病突变之间的明显对应关系。在这些蛋白质中,与结构损伤相关并且与疾病有关的突变残基通常具有更高的TIF值,而中性突变则更多分布在拓扑贡献较低的区域。类似现象在其他蛋白质中也可以观察到,例如人类腺苷琥珀酸裂解酶以及CFTR蛋白,并在整个数据集中表现出一致趋势。一维拓扑特征分析也得到类似结果。
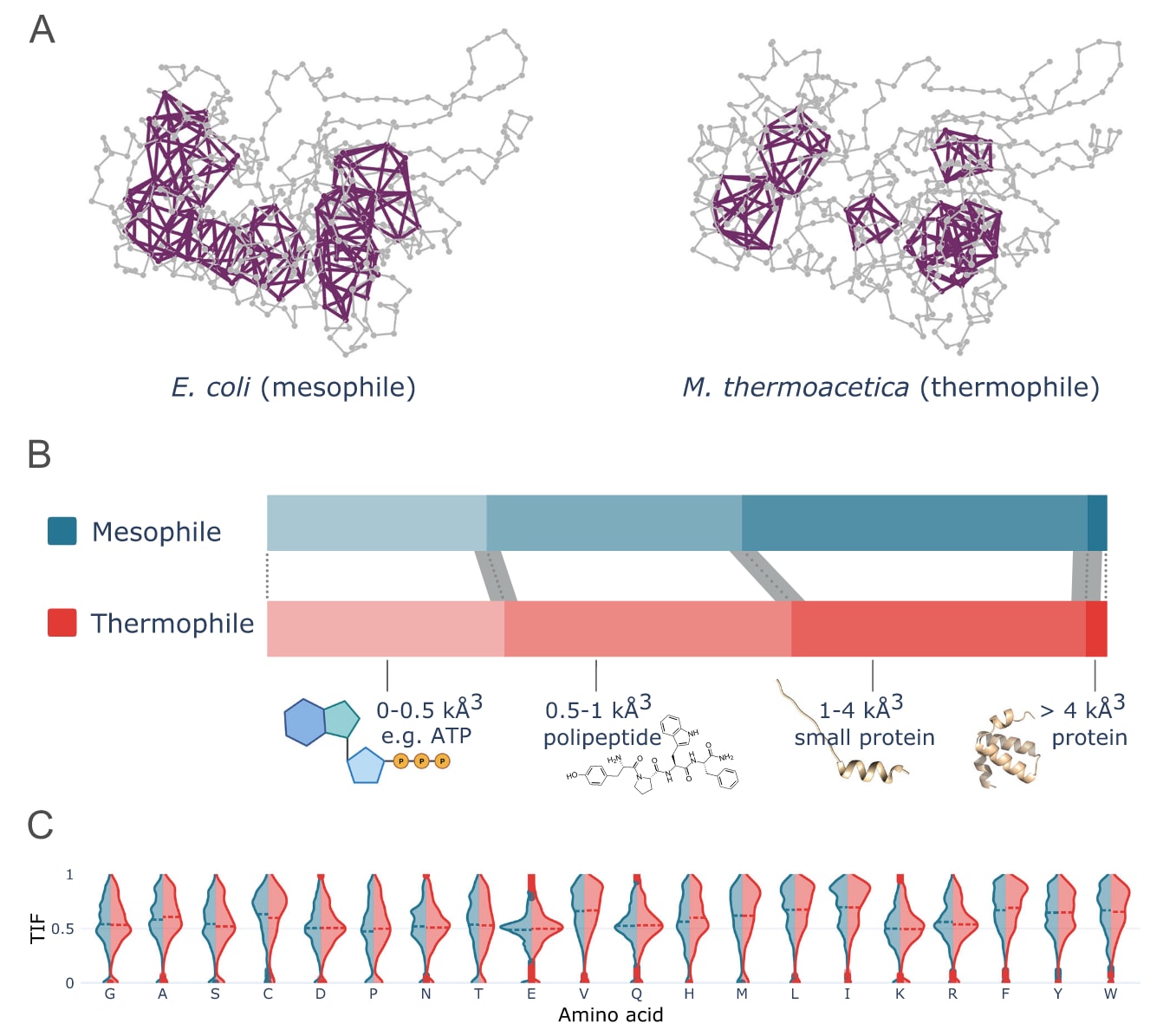
图 4 | 嗜热与中温蛋白质在拓扑上存在差异。 在二维空间中,高度持久的拓扑特征表现为蛋白质结构中的空腔。嗜热生物中的空腔体积一般小于中温生物中的对应空腔,以葡萄糖-6-磷酸-1-脱氢酶为例加以说明(A)。这一现象在来自 10 个不同酶学委员会编号(EC 编号)的酶中普遍存在,并以堆叠式水平条形图展示(B)。因此,每个分箱的面积对应一定体积范围内空腔所占的比例,四个分箱均配有体积具代表性的分子示意图。分箱边界(虚线)处的灰色误差棒为对 1000 个空腔采样所得的标准差。此外,未检测到空腔周围氨基酸频率的补偿性变化足以削弱上述结果的显著性。针对每种氨基酸(按体积从小到大排列),以拓扑影响分数(TIF)分布展示其在空腔周围的占据情况(C),虚线表示平均值。ATP 与多肽图标由 BioRender 制作。源数据提供于 Source_data/source_data_Figure4C-S16.tsv。
3 讨论
研究表明,拓扑方法可以作为解释AlphaFold2结构数据的重要工具。构建的分析流程能够在时间和计算成本可控的情况下,对全部2.14亿个预测蛋白质结构进行拓扑分析,从而揭示蛋白质宇宙中新的整体规律。通过多个应用实例展示了这一方法的价值,包括利用拓扑分析研究蛋白质结构域和结合位点等宏观结构特征,比较嗜热与中温蛋白之间的结构差异,以及分析致病突变对蛋白质结构的影响。
为了方便研究社区使用这些数据,相关研究提供了一个约20TB规模的在线资源,其中包含所有一维和二维持久图、拓扑特征以及逐残基的TIF值。整体分析表明,拓扑方法能够帮助理解庞大的蛋白质结构数据。值得注意的是,所有分析仅基于AlphaFold2提供的Cα原子空间位置(以及少量PDB实验结构用于验证),并未使用任何额外的生物学信息,例如序列信息。因此,在未来研究中,如果进一步整合氨基酸的生物物理和生化性质以及更精细的三维结构信息,可能揭示更多影响蛋白质功能的重要因素。目前的研究已经表明,拓扑特征为蛋白质功能预测和结构生物物理分析提供了新的维度。
尽管拓扑信息本身可能不足以完全解释或设计蛋白质功能,但这一方法为理解AlphaFold2所产生的海量结构数据提供了一条自然且直接的途径,并有望促进生命分子机器的功能与进化研究。未来一个值得探索的方向是将二级结构信息整合进拓扑分析框架中。已有研究提出将持久同调方法与二级结构甚至原子级细节结合的策略。然而,尽管在已解析结构中二级结构注释已十分成熟,但对于仅有序列信息的蛋白质而言,二级结构仍需预测,因此AlphaFold2预测结果的不确定性在一定程度上会给进一步的数据预处理带来挑战。