Nat. Methods 2025 | Foldseek-Multimer:实现快速而灵敏的蛋白质复合物比对
今天介绍的这项工作来自 Nature Methods。该文章围绕蛋白质复合物结构比对在大规模数据时代所面临的计算瓶颈展开,提出并验证了一种全新的解决方案Foldseek-Multimer。随着AlphaFold-Multimer等预测工具的普及,复合物结构的数量正迅速膨胀,但现有比对方法在速度或灵敏度上难以同时满足需求,限制了这些结构在功能注释与进化分析中的应用。作者的核心贡献在于将复合物比对问题转化为**“链对链比对+空间叠合一致性判定”的几何问题**,通过对链级叠合向量进行高效聚类,快速识别能够共同定义复合物对齐的链组合。这一思路避免了对所有链配对方案的穷举搜索,在算法层面显著降低了计算复杂度。在系统基准测试与真实生物学案例中,Foldseek-Multimer表现出与当前参考方法几乎相当的比对质量,同时在速度上提升了数个数量级,使得在数小时内完成数百亿对复合物的比较成为可能。尤其值得注意的是,该方法在低序列相似性条件下仍保持较高灵敏度,为发现“序列不相似但结构保守”的复合物关系提供了有效工具。整体而言,这项工作不仅提出了一种高效的复合物结构比对算法,也为在AlphaFold时代系统性挖掘蛋白质复合物结构空间奠定了方法学基础,对结构生物信息学和大规模功能推断具有重要意义。

获取详情及资源:
- 📄 论文: https://www.nature.com/articles/s41592-025-02593-7
- 💻 代码: https://github.com/steineggerlab/foldseek/
0 摘要
计算结构预测技术的进步将极大地扩充当前已获得的数十万个蛋白质复合物结构。要将这些结构转化为科学发现,关键在于对它们进行结构比对,但这一过程在计算上代价高昂。Foldseek-Multimer通过识别彼此兼容的链对链比对来计算复合物层面的结构对齐,这些兼容关系是通过对其空间叠合向量进行高效聚类得到的。在保持与当前参考方法相当的比对质量的同时,Foldseek-Multimer在速度上快了3到4个数量级,使其能够在11小时内比较数十亿对蛋白质复合物。Foldseek-Multimer以开源软件形式提供,可通过GitHub、在线搜索服务器以及BFMD数据库获取。
1 引言
两个蛋白质复合物之间的相似性体现在其最优的结构比对中,而这一比对结果同时决定了复合物中各条链之间的对应关系。对四级结构进行比对与比较,对于量化复合物的结构多样性、识别不同构象或同源体之间的结构相似性与变化至关重要。此外,由于许多蛋白质是以复合物形式发挥功能的,复合物结构比对也是理解蛋白质功能的重要基础。
近年来,Foldseek被开发为一种快速的结构比对工具,用于检测单链蛋白之间的相似性。该方法通过3Di这一专门描述三级氨基酸相互作用的字母表来表示蛋白质结构,从而能够在大型数据库中高效搜索单链结构相似性,例如在AlphaFold蛋白质结构数据库中。然而,由于复合物比对需要事先确定不同链之间的正确配对关系,Foldseek本身无法直接用于两个复合物之间的结构对齐。
US-align是一种通用的结构比对工具,可用于多种分子类型,包括蛋白质复合物。其复合物比对策略以TM-score最大化为目标。由于链配对的可能组合数量呈阶乘增长,US-align采用贪婪搜索启发式方法提出候选链配对方案,再通过动态规划进行细化。实践表明,这一策略使US-align在保持更高比对得分的同时,比此前的MM-align快最多五倍,因此被视为成对复合物结构比对的参考标准。
为了在大型数据库中发现结构上保守的界面配对,Dey等人提出了QSalign,用于检测相似的同源多聚体复合物。QSalign通过基于序列相似性进行预筛选,仅对序列一致性约为25%或更高的复合物对执行完整的结构比对,从而节省了计算时间。这种加速是以牺牲灵敏度为代价的,使其难以发现位于所谓“暮光区”或更低序列相似性范围内的结构相似复合物。即便如此,在使用100个线程的条件下,QSalign对约十万个复合物组成的3DComplex数据库进行全对全搜索仍然耗时数月。
为进一步降低数据库搜索中的计算成本,Guzenko等人提出了一种替代方法,通过3D Zernike描述符比较两个复合物的整体形状,从而避免了链配对过程。这一方法能够在不到一秒的时间内查询数十万个结构。然而,由于其仅基于整体形状相似性进行匹配,只能发现全局形状相近的分子,相较于US-align和QSalign等基于链配对的方法,其灵敏度受到限制。此外,该方法也无法识别那些在整体上不匹配、但在局部链区域存在相似性的情况。
随着利用AlphaFold-Multimer等工具对蛋白质复合物进行计算预测已能够在全蛋白组尺度上系统性地开展,并进一步应用于宏基因组样本序列,对大型数据库进行高灵敏度搜索的挑战将愈发突出。这一趋势预计将在未来几年内为结构数据库带来数量庞大、甚至可能达到数百万级别的复合物结构。
为满足复合物之间大规模结构比较的需求,提出了Foldseek-Multimer这一方法 (图1)。其高效性主要来源于三个方面:首先,利用Foldseek实现快速的链对链结构比较;其次,将链对链比对表示为空间叠合向量,并通过高效聚类从中识别复合物层面的结构对齐;最后,在搜索过程中使用聚类后的数据库以减少冗余计算。基准测试结果表明,Foldseek-Multimer在准确性上与US-align几乎相当,但在速度上提升了多个数量级;同时,该方法在低序列相似性的条件下仍保持较高灵敏度,适用于宏基因组来源的复合物研究;此外,它还能够执行全对全搜索,在11小时内比较数十亿对蛋白质复合物。
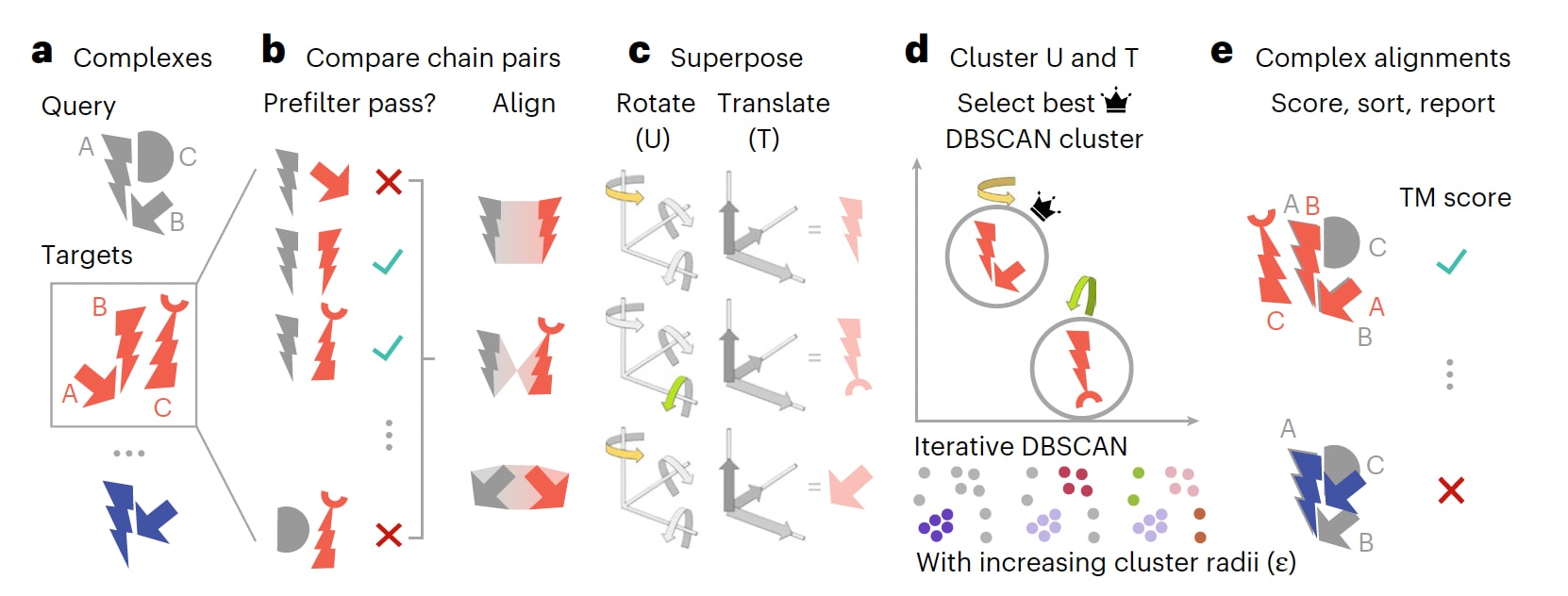
**图1|Foldseek-Multimer的整体示意图。 **a,Foldseek-Multimer支持将一个或多个查询复合物快速检索至大型结构数据库中,该数据库可包含数百万个目标复合物。b,查询复合物中的所有链以灰色表示,与每一个目标复合物中的链以红色表示进行两两比较。通过预过滤步骤,可迅速剔除明显不匹配的链对,从而仅对具有潜在相似性的复合物对执行完整结构比对。c,Foldseek-Multimer将每一次链对链比对表示为一个空间叠合,由将目标链叠合到查询链所需的旋转和平移参数描述。在这一简化示例中,上方和下方的两个链对链比对对应于沿同一轴的旋转,而中间的比对则对应于沿另一轴的旋转。d,复合物层面的结构对齐由链对链比对推断而来,其依据是属于同一复合物对齐的链对在空间叠合上具有相似性。Foldseek-Multimer通过逐步增大半径、迭代地应用DBSCAN算法来识别叠合向量的聚类,并选取得分最高且有效的聚类用于计算复合物对齐。e,基于得分最高的聚类结果,在查询复合物与目标复合物之间的所有链对齐基础上计算整体的复合物TM-score。
在一个包含931对已知结构相似蛋白质复合物的基准数据集上,对Foldseek-Multimer与US-align的比对质量进行了系统比较。Foldseek-Multimer分别在两种运行模式下进行测试,两种模式在链对链比对算法上有所不同,分别采用3Di+AA编码的Foldseek-MM模式,以及基于TM-align的Foldseek-MM-TM模式。结果显示,三种方法均能将绝大多数复合物对判定为结构相似,当采用
对各工具的运行时间进行了系统测量,并进一步分析了Foldseek-Multimer各组成部分对整体加速效果的贡献。首先,在计算931对复合物成对比对这一任务中,相较于US-align观察到1到2个数量级的速度提升 (图2b),这一结果反映了链对链比对阶段(Foldseek-MM)以及叠合向量聚类阶段(Foldseek-MM和Foldseek-MM-TM)的高效性。尤其是Foldseek-MM-TM的表现,凸显了利用空间叠合作为替代全局结构比对策略这一设计在Foldseek-Multimer中的关键作用。随后,以基准数据集中涉及的677个复合物为查询,对3DComplexV7数据库进行了搜索。在这一数据库检索场景中,由于额外引入了预过滤步骤,Foldseek-Multimer相较US-align实现了3到4个数量级的进一步加速。
在具体应用示例中,Altae-Tran等人此前在来源于Sulfitobacter sp. JL08的环境样本中发现了首个具有明确干扰机制的CRISPR–Cas IV-A系统。基于这一发现,研究利用ColabFold-AlphaFold-Multimer对其核糖核蛋白复合物的一部分结构进行了预测,该预测结果的质量处于可接受水平,其
除核心算法本身的高效性外,Foldseek-Multimer在该场景中还得益于PDB100作为聚类数据库的结构设计,使其仅需先搜索343,785个代表结构,并只在具有潜力的结构簇中展开进一步搜索,从而获得了额外的加速效果。在搜索结果中,Foldseek-MM-TM和US-align均给出了5个
随后,在全对全比对的设置下对Foldseek-Multimer进行了进一步评估,所使用的是此前已通过QSalign分析过的3DComplexV7数据库。QSalign依赖计算代价较高的Kpax结构比对方法,因此无法执行穷尽式的结构搜索。为降低计算量,QSalign首先基于序列相似性筛选出约5800万个复合物对,随后仅对这些候选对应用Kpax,最终识别出约450万个结构相似的同源多聚体复合物对,即所谓的QSalign配对。
在使用128个计算核心的条件下,Foldseek-MM对经过聚类处理的3DComplexV7数据库进行了自比对,在11小时内检查了约570亿个复合物对。在采用与QSalign相同的
除命令行工具外,Foldseek-Multimer还被集成至Foldseek在线服务器,并通过NGL Viewer库对搜索结果进行可视化展示。服务器以半透明着色的蛋白质表面叠加方式呈现链对链配对关系,用户可以在不同的比对模式之间切换,并通过分类学过滤条件将搜索范围限定在特定进化类群中。为配合在线服务中对预测结构的检索,整合了来自多个社区项目的297,570个多聚体结构预测,构建了统一的BFMD数据库。该数据库既可通过在线服务器访问,也支持本地使用。
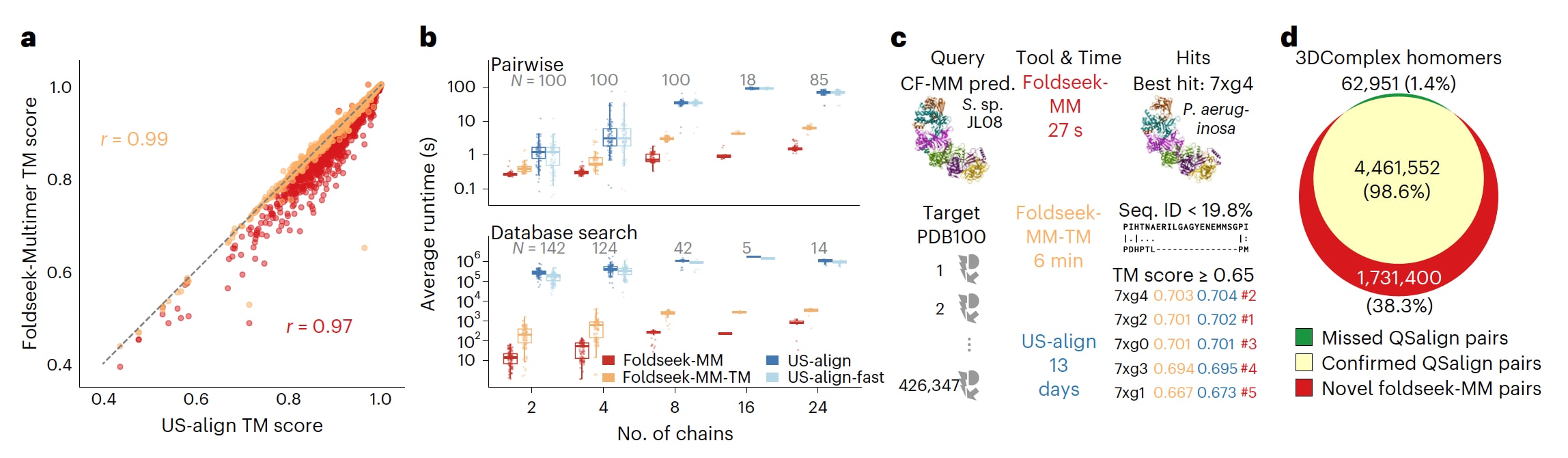
图2|Foldseek-Multimer的性能评估。 a,在931对已知结构相似的蛋白质复合物上,分别使用US-align和Foldseek-Multimer计算得到以查询长度归一化的TM-score,其以目标长度归一化的结果见补充图。两种方法计算得到的TM-score之间具有高度相关性,皮尔逊相关系数接近1。b,基于与a中相同的数据集,对工具的运行时间进行了比较。复合物按所包含的链数进行分组,图中展示了部分代表性分组。箱线图显示了四分位数分布,每一个点代表一个复合物对(上)或一个复合物(下),样本数量以N标注,触须延伸至距离上四分位数或下四分位数1.5倍四分位距范围内的最大或最小值。在成对比对模式下,由于高效的链对链比对与叠合向量聚类,Foldseek-Multimer的速度比US-align快10到100倍。在数据库搜索模式下,查询对象为3DComplexV7数据库,得益于预过滤步骤的进一步加速,Foldseek-Multimer的速度提升达到
2 结论
总而言之,该工作提出了一种用于复合物对复合物结构比对的策略,通过链对链比对所对应的空间叠合关系,快速识别彼此兼容的比对集合。尽管该研究以蛋白质复合物为例进行了展示,但在能够对单个亚基进行比对的前提下,Foldseek-Multimer的策略同样具有推广到RNA或DNA复合物结构的潜力。凭借前所未有的灵敏度与计算速度,Foldseek-Multimer成为AlphaFold时代研究大规模复合物结构不可或缺的工具。