Nat. Methods 2025 | Scikit-bio:用于生物组学数据分析的基础性Python库
今天介绍的这项工作来自 Nature Methods。组学数据已成为现代生命科学研究的核心数据形态,在揭示复杂生物系统方面具有不可替代的价值。然而,这类数据天然具有高维、稀疏和组成性等统计特征,使得通用数据分析方法往往难以胜任。如何在计算层面充分利用组学数据中蕴含的生物学结构信息,成为长期存在的关键问题。该文介绍了scikit-bio这一面向组学数据分析的Python基础库。该库围绕样本×特征数据表及其相关树结构展开设计,系统性地整合了稀疏数据表示、组成性变换、多样性度量、基于距离的统计分析以及系统发育信息建模等核心能力。通过与Python科学计算生态的深度兼容,scikit-bio在传统生物信息学方法与现代机器学习框架之间建立了有效连接。该文章不仅阐述了scikit-bio在方法论与软件工程层面的设计思想,也回顾了其在微生物组学及其他组学领域中的应用演进。作为一款社区驱动、长期维护的基础性工具,scikit-bio展示了面向组学数据分析的软件应如何在生物学可解释性、计算效率与可扩展性之间取得平衡。

获取详情及资源:
- 📄 论文: https://www.nature.com/articles/s41592-025-02981-z
- 💻 代码: https://github.com/scikit-bio/scikit-bio
0 引言
现代生物学研究通常以多种“组学”数据为核心特征,这些数据用于描述生物实体的整体状态,包括基因组学、转录组学、蛋白质组学、代谢组学和宏基因组学等。组学数据为理解复杂生物系统提供了前所未有的视角,但同时也带来了长期存在的分析难题,例如高维性(特征数量远多于样本数量)、稀疏性(大多数特征值为零)以及组成性(样本内特征之间相互依赖)。尽管如此,组学数据也蕴含着独特的分析潜力,其生物学特征往往通过知识驱动的结构相互关联,并常以树状图的形式存在,如系统发育树或功能分类体系。这类结构信息可以被有效利用以增强分析能力,而这些特性也使得通用数据分析方法在组学研究中显得力不从心。
为应对上述需求,提出了scikit-bio这一面向组学数据分析的Python生物信息学库。该库包含500余个对外接口函数、类和方法,构建了一套系统而完整的数据结构与算法体系,专门用于解决组学研究中固有的分析挑战与机遇。尽管scikit-bio也支持基础的序列分析功能,其核心优势在于对样本×特征数据表及其相关树结构的分析,这两类数据表示形式在多数组学研究中最为常见。这一设计取向使其区别于BioPython等通用工具包,以及ScanPy等面向特定领域的软件框架。同时,scikit-bio在以R语言为代表的传统组学分析方法与Python生态中广泛使用的机器学习框架之间发挥了连接作用。
在应对组学数据稀疏性方面,scikit-bio整合了生物观测矩阵(BIOM)格式,这是一种面向表格型数据的稀疏矩阵结构,并已成为微生物组研究中的通用标准。在处理数据组成性问题时,该库提供了多种变换方法,包括中心化对数比变换(CLR)以及用于零值处理的乘法替换方法,同时实现了考虑组成性的差异丰度检验方法,如ANCOM。除此之外,scikit-bio还提供了多种用于量化样本内与样本间生物多样性的指标。样本间多样性分析通常生成距离矩阵,这些结果可进一步用于主坐标分析(PCoA)等多维尺度变换方法,或用于置换多元方差分析(PERMANOVA)等多变量统计检验,以评估组学特征与表型之间的关联关系。此外,该库还实现了Mantel检验,用于评估不同组学数据之间的相关性。
scikit-bio同时提供了丰富的树结构操作功能,用于刻画特征之间的生物学相关性。使用者可以构建、遍历和操作树结构,计算特征或特征集合之间的距离,并将树信息整合进群落建模分析中,例如Faith系统发育多样性指标和UniFrac距离。这种方法能够将生物学先验知识引入高维数据分析过程,使原本在计算上难以处理的问题变得可分析。
该库中的多项实现方案在Python生态系统中处于先进水平,部分功能具有独特性,并针对当代及新兴研究中的超大规模数据分析需求进行了优化,例如可支持样本数量超过100,000的PCoA分析。
在软件架构层面,scikit-bio与Python科学计算生态系统实现了高度集成。其结构化API遵循现代设计与风格规范,并广泛采用NumPy数组、SciPy稀疏矩阵和Pandas数据框等高效数据结构,使得已有数据能够直接用于分析而无需进行代价高昂的格式转换。分析结果也可直接传递给其他常用库,用于统计建模、机器学习或数据可视化。同时,其灵活且可扩展的输入输出系统使其能够与非Python环境下的生物信息学工作流程协同使用。
自2014年首次发布以来,scikit-bio已支持了大量研究工作和分析工具的开发,其中包括对广泛使用的微生物组数据分析平台QIIME多个关键功能的支撑。尽管该项目起源于微生物组研究,其整体设计基于组学数据的共性特征,因此具备良好的通用性。作为一个独立的软件库,scikit-bio采用模块化架构,允许使用者按需选择和组合功能模块,并将其嵌入到特定领域的分析流程中。目前,其应用范围已扩展至单细胞数据分析、结构变异分析、代谢组学和表观遗传学等多个研究方向,相关成果已被引用数万次。
在软件质量保障方面,项目遵循工业级开发规范,单元测试覆盖率达到98%,并配备完善的文档测试和风格约束机制。在线文档系统详细介绍了相关方法的数学背景、生物学应用场景及示例代码,同时提供围绕模块与工作流程展开的结构化教程。此外,scikit-bio还被用于支撑一本生物信息学教材,有效降低了使用门槛并促进了跨学科合作。
scikit-bio采用社区驱动的发展模式,迄今已有80余名开发者参与贡献。这种模式保证了软件能够随着科学计算领域的发展持续演进。该项目致力于不断改进和扩展功能,以支持更前沿、更广泛的研究应用,并被视为未来仍将长期服务科研社区的一项关键基础性软件工具。
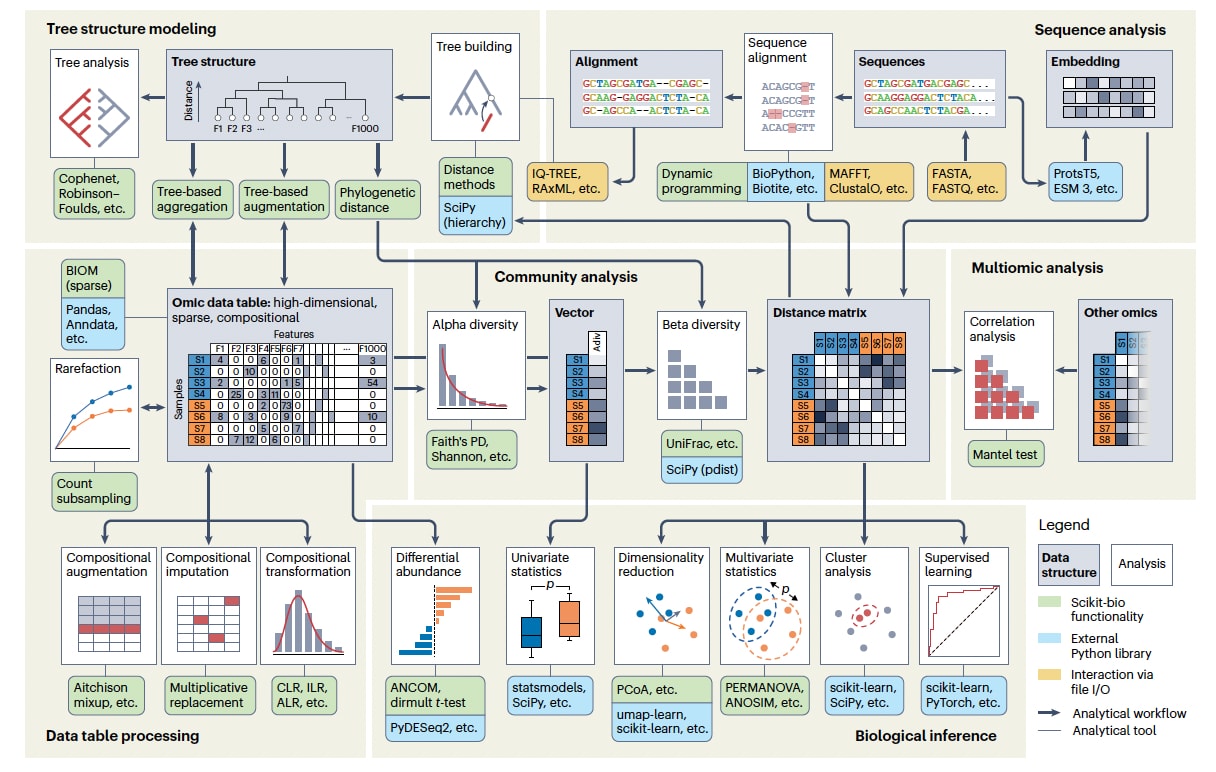
**图1|scikit-bio在生物组学数据分析中的能力以及其与Python科学计算生态系统中其他工具的交互关系。**带直角边框并包含示意图的方框表示数据结构(浅灰色背景)或分析过程(白色背景)。带圆角的方框表示具体的分析工具,并按来源分为三类:绿色表示由scikit-bio实现的功能;蓝色表示由外部Python库提供、可在Python框架内与scikit-bio协同使用的功能,例如由scikit-bio生成的距离矩阵可以输入SciPy用于层次聚类,输入scikit-learn用于