Structure 2025 | 重新思考pLDDT在蛋白质柔性研究中真正揭示了什么
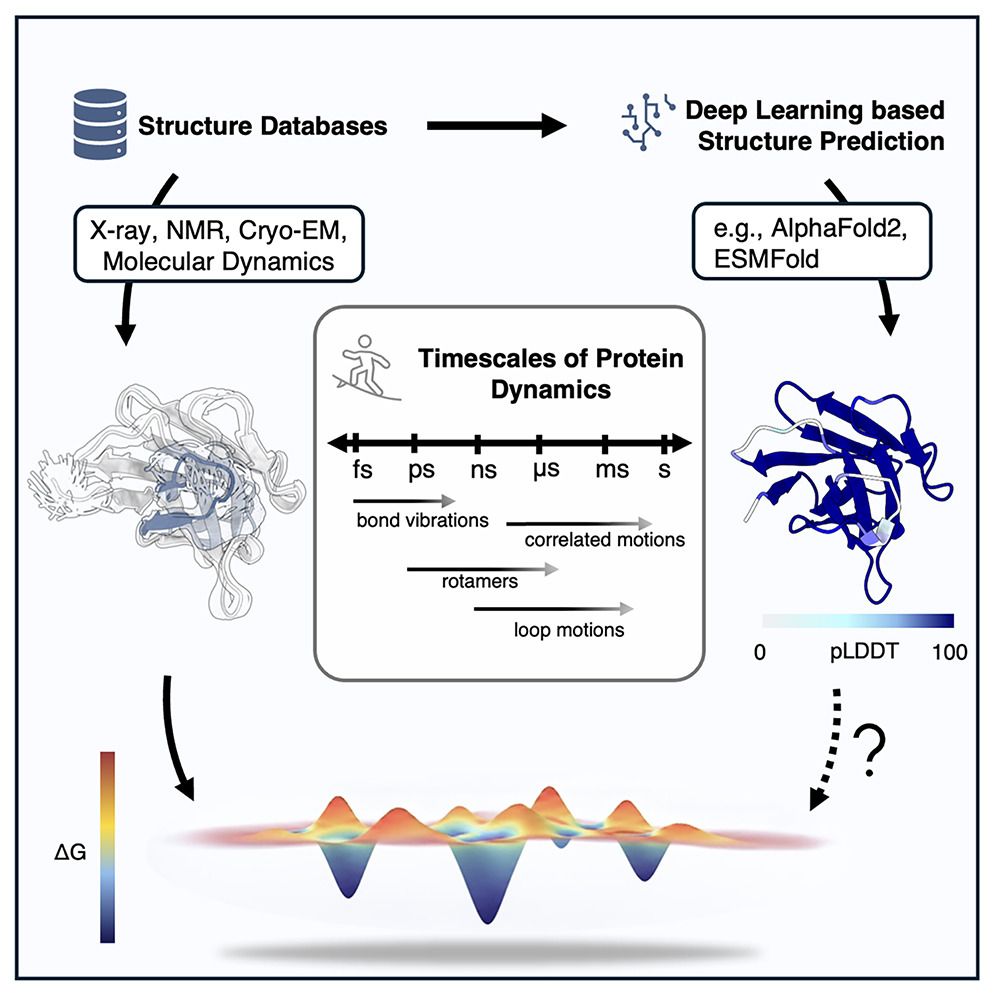
今天介绍的这项工作来自 Structure。近年来,以AlphaFold2为代表的深度学习方法极大改变了蛋白质结构预测领域,pLDDT作为常用的置信度指标,也逐渐被一些研究解读为蛋白质柔性的替代量。然而,pLDDT究竟能在多大程度上反映蛋白质真实的构象变化与动力学行为,仍然存在明显争议。
该文围绕这一问题,系统讨论了pLDDT的物理含义及其固有局限,强调pLDDT本质上衡量的是模型对单一结构假设的确定性,而非蛋白质在不同时间尺度上的构象采样能力。通过结合分子动力学模拟、核磁共振、X射线晶体学与冷冻电镜等方法的研究现状,文章指出,高pLDDT并不等同于结构刚性,功能相关区域即便在预测中呈现高置信度,仍可能具有显著的构象可塑性。
在此基础上,文章进一步评述了利用AlphaFold架构与生成式深度学习模型探索构象集合的最新进展,并指出现有训练数据在时间尺度和构象多样性上的不足。作者强调,构建更加系统、覆盖微秒至毫秒尺度的动力学数据集,是推动蛋白质构象能景预测与功能理解的关键一步。整体而言,该文为如何理性解读pLDDT,以及如何将人工智能结构预测与蛋白质动力学研究更有效地结合,提供了重要的思考框架。

获取详情及资源:
0 摘要
深度学习模型通过实现对蛋白质三维结构的可靠预测,并提供如预测局部距离差检验值(pLDDT)等置信度指标以估计局部不确定性,从而深刻改变了结构生物学领域。然而,pLDDT是否能够反映蛋白质的本征柔性,目前仍不清楚。通过实验与计算方法对蛋白质柔性与动力学进行定义和量化,对于提升在不同时间尺度上对构象变化的建模与解释能力至关重要。
1 引言
与均方根偏差(RMSD)等依赖结构比对的指标不同,局部距离差检验(LDDT)最早于2013年提出,是一种不依赖结构比对的评分方法,通过评估预测结构模型内部原子间距离来衡量结构质量。随着AlphaFold2(AF2)的发布,以及ESMFold、OpenFold和RosettaFold等深度学习结构预测方法的不断涌现,预测结构模型的置信度通常通过预测局部距离差检验值(pLDDT)进行量化。由于该指标刻画的是局部结构不确定性,其作为蛋白质柔性替代指标的潜在用途已成为多项研究关注的焦点。然而,pLDDT的一个明显缺陷在于,它并未包含任何关于动力学时间尺度的信息。
评估蛋白质柔性对于理解蛋白功能和基本生物学机制至关重要,因为许多生物学功能依赖于不同构象状态之间的转变。多种实验和计算技术能够捕捉从皮秒到毫秒尺度的构象变化与蛋白质动力学行为。蛋白质运动往往比简单的两态相互转化更为复杂,快速的皮秒至纳秒级运动可以与更慢的微秒至毫秒级重排相互耦合,甚至诱发后者,共同塑造多维度的构象能景。需要指出的是,结构位移的幅度例如RMSD,并不必然与构象状态的时间尺度或占据比例相关。传统上,蛋白质柔性,或者说皮秒至纳秒时间尺度上的各向同性运动,通常可以通过核磁共振谱学、X射线晶体学的构象集合精修、冷冻电镜的三维变异分析以及分子动力学模拟进行表征。
为评估pLDDT与蛋白质柔性之间的关系,在本期Structure中,Vander Meersche等人开展了大规模分析,并将结果与公开数据集进行比较。该研究整合了多种结构变异来源,包括ATLAS数据集中的分子动力学轨迹、来自PDB的核磁共振不确定性结构集合以及晶体学B因子,并将这些结果与AlphaFold2和ESMFold给出的逐残基pLDDT分数进行对照。然而,在不同方法之间对“柔性”的整体解读仍需保持谨慎,因为这些替代指标混合了本质上不同的现象,例如力场驱动轨迹中的构象采样、由数据稀疏性导致的不确定性,以及X射线晶体学B因子中同时包含的热运动和模型不确定性。
在涉及配体结合或变构调控等功能过程的区域中,尽管实验或计算结果表明存在构象变异,AlphaFold2往往仍预测出整体均一且较高的pLDDT值。这一现象已在多个体系中被独立观察到。例如,已有研究表明,已知在不同功能状态之间切换的转运蛋白和G蛋白偶联受体,默认情况下通常被预测为单一的高置信度构象。类似地,Vander Meersche等人报道,在大肠杆菌有机汞裂解酶体系中,尽管计算和实验结果均提示配体结合位点在游离态下发生环区重排并采样多种构象,AlphaFold2仍预测出结合态构象,且pLDDT整体较高。这种现象并不令人意外,因为pLDDT本质上是对模型不确定性的置信度度量,而非蛋白质柔性或动力学行为的直接指标。
这一局限源于基于机器学习的结构预测方法所固有的约束,其训练数据主要由实验解析得到的结构模型构成,而这些模型在很大程度上排除了动态构象或低概率状态。因此,pLDDT值反映的是模型对某一单一结构假设的内部确定性,而非蛋白质真实存在的构象能景。尽管已有研究表明,pLDDT与皮秒至纳秒尺度的运动存在一定相关性,但反过来并不成立,即高pLDDT并不意味着结构刚性。某一残基在特定构象中的取向可以被高度确定地预测,同时仍然具备重排并转变至其他构象状态的能力。模型对某一构象状态的置信度还可能受到序列保守性、模板比对不确定性,甚至缺失数据等因素的影响。
已有多项研究聚焦于利用AlphaFold2架构对替代构象状态进行采样。一些方法通过操控输入的多序列比对以削弱进化信号,从而尝试从AlphaFold2中提取构象集合,例如随机子采样、多序列比对聚类、比对列遮蔽或CF-random等策略。以这些子采样得到的结构作为起点开展分子动力学模拟,在探索功能相关的构象集合方面显示出一定潜力。迄今为止,在采样充分的前提下,分子动力学模拟仍然是定量评估构象集合热力学与动力学性质最为可靠的基于物理的计算工具,前提是功能相关的构象状态及其转变能够被充分捕捉。
深度学习方法的迅速发展催生了一系列用于预测蛋白质构象集合的模型与技术,有望弥合人工智能结构预测与蛋白质运动物理建模之间的差距,并推动该领域朝着统一理解结构与动力学的框架迈进。扩散模型或流匹配模型,例如BioEmu和AlphaFlow,通常基于MD-CATH中的ATLAS数据库所包含的分子动力学数据或内部生成的数据集进行训练。需要注意的是,这些数据集中大多数轨迹仅对应于单个蛋白100至200纳秒尺度的模拟。此类数据集能够为局部蛋白质柔性与纳秒尺度波动提供有价值的信息,但相应地,训练得到的模型及其柔性比较也受限于纳秒时间尺度,这很可能限制了其跨越高能垒、刻画蛋白质复杂多维构象能景的能力。
为了推动能够可靠预测完整构象能景并捕捉功能相关微秒至毫秒尺度运动的生成模型发展,该领域迫切需要更加丰富且标准化的分子动力学或实验数据集。已有研究表明,精心构建的数据集对于预测微秒至毫秒尺度的运动至关重要,并正在改变对蛋白质动力学与功能的预测和理解方式。除提升模型精度之外,构建和整理超越静态单一结构的数据集,将为训练提供比当前蛋白质结构数据库中更为丰富和多样的信息。当前正处在深入理解生物大分子热力学能景的重要契机之中,而这一愿景的实现需要持续且广泛的共同投入,不仅能够提升对蛋白质动力学与功能的预测能力,也将加深对生物学功能进行学习与推理的整体认知。