J. Med. Chem. 2025 | 抗体−药物偶联物中肽连接子的最新进展
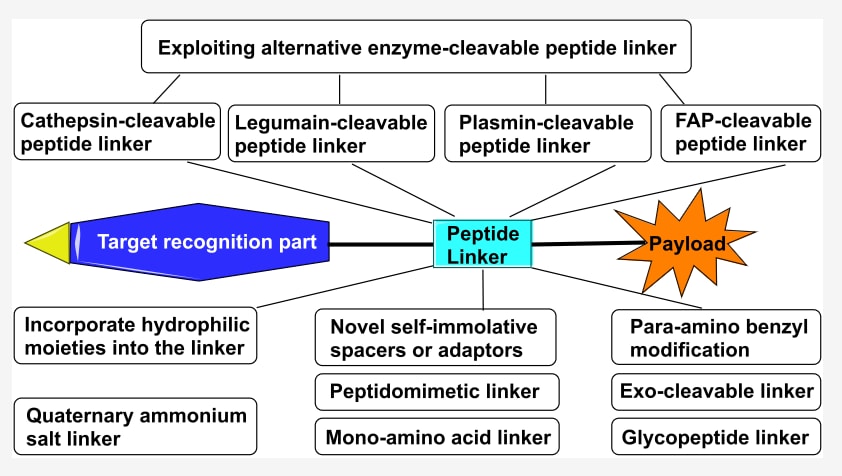
今天介绍的是发表在 《Journal of Medicinal Chemistry》 上的一篇综述,主题聚焦于抗体−药物偶联物(ADCs)中肽连接子的最新进展。文章系统梳理了新型连接子设计如何改善药物递送效率与治疗效果。传统的 cathepsin 可切割肽连接子 已在多种临床获批 ADC 中广泛应用,但仍存在药物过早释放及相关毒性等局限。为此,研究者们探索了替代性酶切肽连接子,以克服这些问题并提高治疗特异性。与此同时,综述还总结了多种 连接子修饰策略,包括在结构中引入亲水性基团、开发新型自裂解间隔子、对对氨基苄基(PAB)进行结构修饰,以及利用季铵盐连接子改善水溶性和降低聚集。这些创新性设计不仅提升了ADC的循环稳定性和抗肿瘤活性,也为未来连接子化学的发展提供了新的方向。
Yang, L.; Ma, J.; Liu, B.; Li, Y.; Ma, Y.; Chen, H.; Han, Z. Recent Advances in Peptide Linkers of Antibody–Drug Conjugates. J. Med. Chem. 2025, 68 (17), 18099–18113. https://doi.org/10.1021/acs.jmedchem.5c01500.
0 摘要
抗体−药物偶联物(ADCs)是一类极具前景的癌症治疗药物。这种创新性的分子设计将抗体的靶向性与延长的半衰期与小分子的细胞毒性巧妙结合,从而实现药物在癌细胞中的选择性递送。连接子分子在ADC疗效中发挥着关键作用。尽管ADC连接子可以分为可切割型和不可切割型,但目前多数获批的ADC均采用可切割型肽连接子。这类连接子可被组织蛋白酶、纤溶酶或legumain等酶切割,既能保证ADC在循环系统中的稳定性,又能在肿瘤内实现细胞毒性载荷的选择性释放。因此,连接子化学已成为ADC研发中至关重要的组成部分。该综述将阐述肽连接子在ADC开发中的作用,重点介绍其最新进展,并对未来的连接子设计方向提出展望。
1 引言
抗体−药物偶联物(ADCs)是一类强效的生物制药分子,能够将高毒性的药物或载荷特异性递送至癌细胞。通常,ADCs的靶向部分为单克隆抗体,而载荷为小分子细胞毒药物,两者通过共价化学连接子结合在一起(Figure 1)。这一创新性设计在血液恶性肿瘤与实体瘤的治疗中均展现出显著疗效。除传统ADCs外,还衍生出多种形式,如双特异性ADCs、前体抗体−药物偶联物(probody−drug conjugates)、抗体片段−药物偶联物(FDCs)、肽−药物偶联物以及小分子−药物偶联物(SMDCs),它们主要区别在于靶向部分。此外,载荷的类型也更为多样,可以是免疫刺激剂、蛋白降解剂、光敏染料、放射性核素或寡核苷酸,从而实现不同的细胞毒性机制。
传统ADCs的作用机制始于抗体与细胞表面相应抗原的结合,随后ADC−抗原复合物被内吞进入内体。早期内体逐渐成熟为晚期内体,最终与溶酶体融合。在溶酶体中,ADC被降解并释放细胞毒性载荷。这些载荷通过与DNA嵌合、作用于线粒体或微管来诱导癌细胞死亡(Figure 2)。部分载荷还可扩散至邻近细胞,产生旁观者效应。值得注意的是,这种效应在某些情况下甚至可独立于内吞发生。在整个过程中,连接子对载荷释放起到至关重要的调控作用,其特性直接决定ADCs的疗效与毒性。
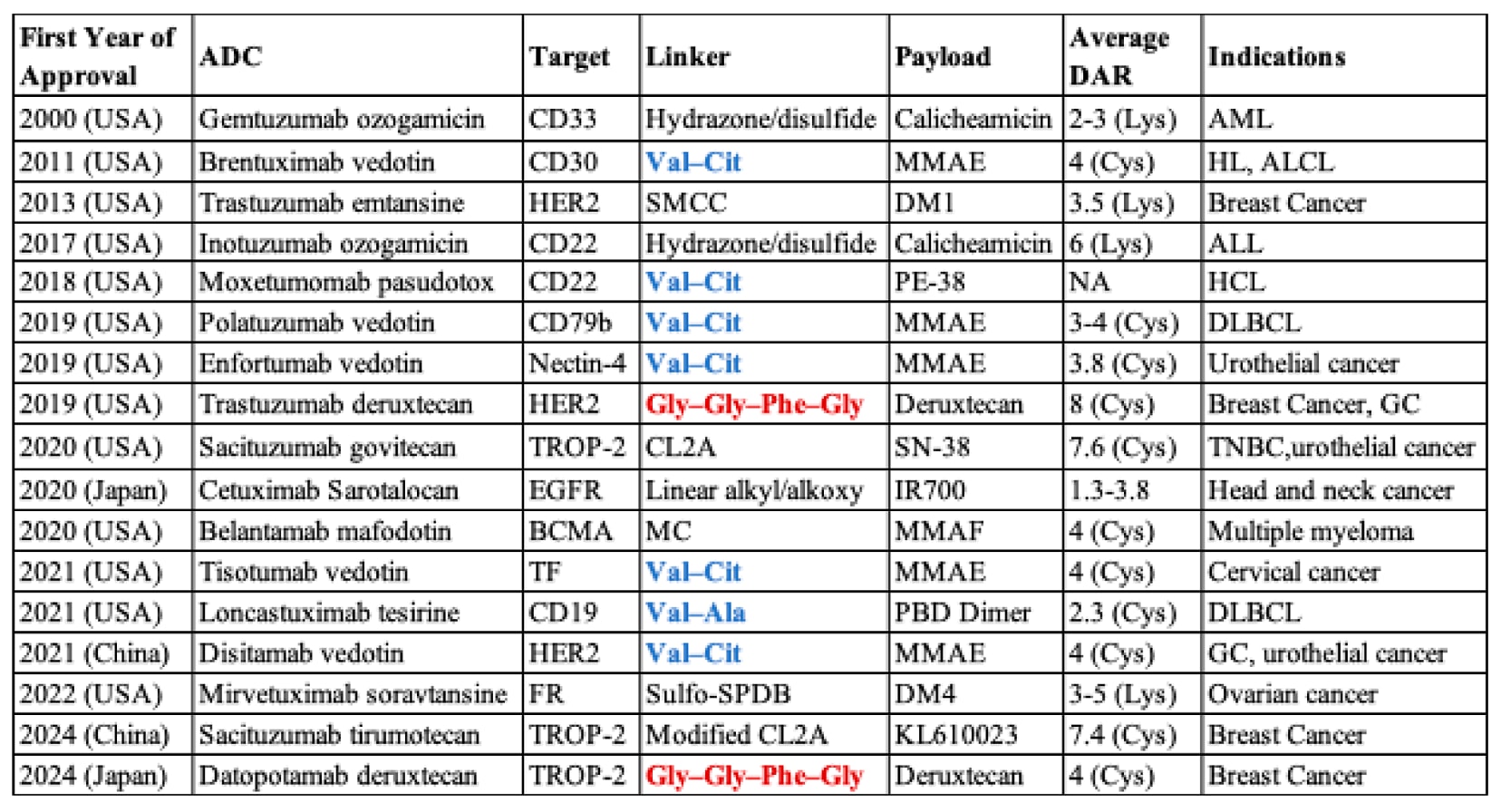
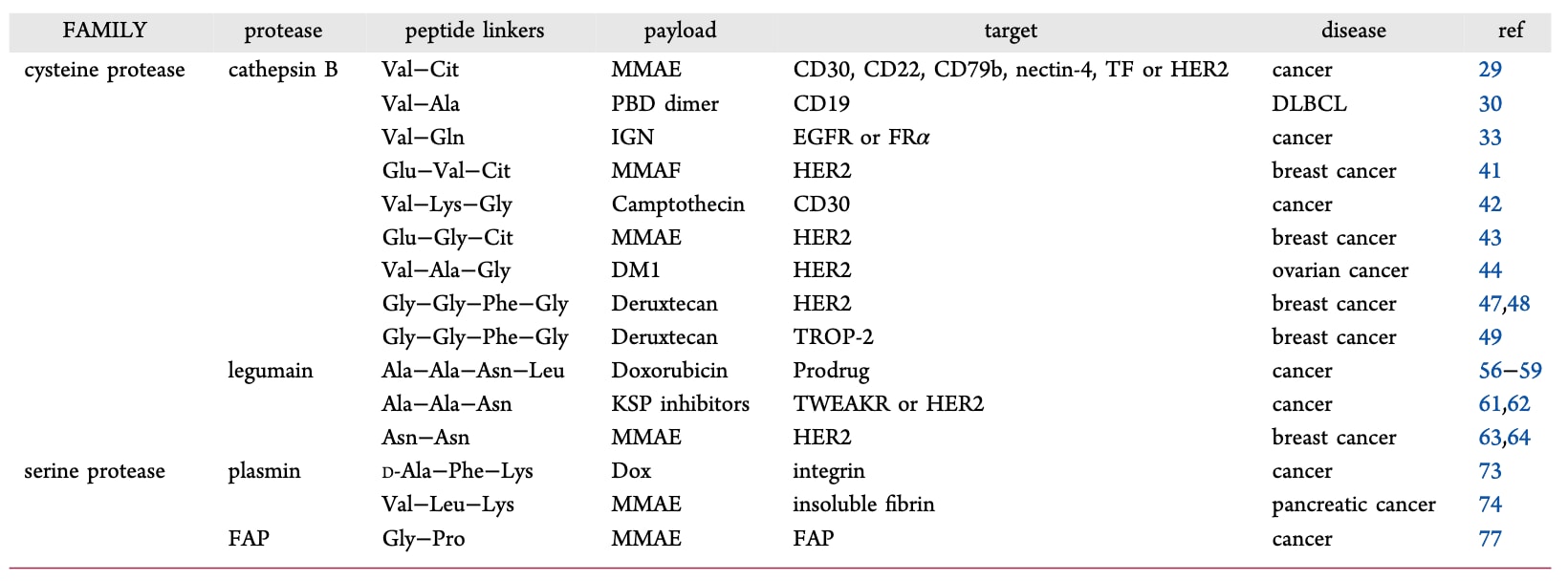
目前,已有17种ADCs获得批准并在全球上市(Table 1),超过100种在临床试验阶段,涵盖20余种血液恶性肿瘤与实体瘤适应症。血液肿瘤靶点包括CD19、CD22、CD30、CD33、CD79b以及B细胞成熟抗原;实体瘤靶点则涉及HER2、nectin-4、肿瘤相关钙信号转导蛋白2、组织因子(TF)与叶酸受体等。ADCs常用载荷包括微管结合剂(如MMAE、DM1、DM4)、DNA靶向药物(如calicheamicin)以及拓扑异构酶抑制剂(如SN-38)。多数获批ADCs采用可切割型连接子,如腙键、二硫键或肽连接子。在17种已批准ADCs中,有9种使用肽连接子,其中包括7种二肽连接子(Val−Cit或Val−Ala)与2种四肽连接子(Table 1)。
尽管ADCs在癌症治疗中表现出优异疗效,但仍面临诸多挑战,包括循环中过早释放载荷导致的意外毒性、开发成本高且周期长、以及连接子溶解性有限等。鉴于连接子在药物递送与作用机制中的核心地位,连接子的设计已成为ADCs研究的重要方向。
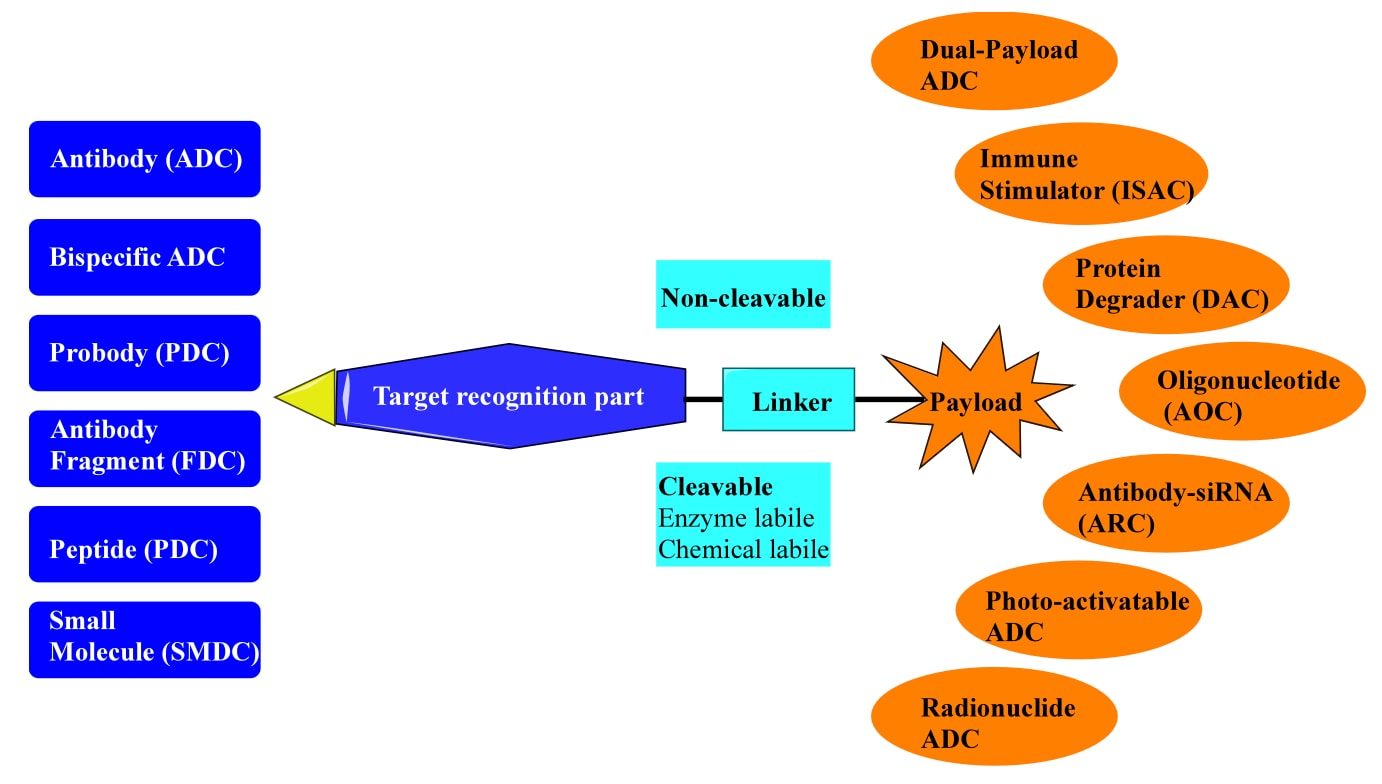
图1 | 基于新型靶点识别或载荷设计的药物偶联物组成与分类。 每一部分——靶点识别单元、连接子和载荷——都在决定药物偶联物性质中发挥关键作用。
2 ADC连接子的类型
ADC连接子分为可切割型与不可切割型两类。根据切割机制,可切割型连接子又可细分为化学切割型与酶切型(Figure 3)。连接子的长度、极性与稳定性对ADC的治疗效果至关重要。连接子的设计通常取决于ADC的用途及作用方式。已有多篇综述总结了不同类型的连接子,因此当前部分主要聚焦于肽连接子,即一类重要的酶切型连接子。作者概述了组织蛋白酶可切割的二肽、三肽与四肽连接子,并重点介绍利用其他酶类作为释放机制的肽连接子开发进展,以及通过连接子修饰提升ADC治疗效果的策略。
2.1 酶切型肽连接子:从前药到ADCs
高效ADC需要在循环中保持载荷−连接子复合物的稳定性,同时在肿瘤细胞内高效释放药物。ADCs通常被转运至肿瘤细胞溶酶体,在此高浓度的水解酶或蛋白酶选择性切割肽连接子,释放载荷。早期的研究主要集中于蒽环类前药,如将阿糖胞苷(DNR)或阿霉素(DOX)与二肽偶联。Ala−Leu−DNR在肿瘤细胞内被激活并水解为DNR。为提升稳定性与切割效率,研究者设计了自裂解型对氨基苄基氨基甲酸酯(PABC)间隔子,并证实其对蛋白酶激活前药设计至关重要。若缺乏PABC间隔子,cathepsin B无法切割前药并释放DOX,ADC也随之失效。
Cathepsin B是一种溶酶体蛋白酶,属于木瓜蛋白酶家族,在大多数肿瘤中过度表达,其活性受限于酸性环境(pH 3.5−6.0),而在正常细胞外环境几乎不表达,仅在病理条件(如转移瘤)下出现。因此,基于cathepsin B可切割的肽连接子通常在循环中保持稳定。研究表明,含有Phe−Lys−PABC−DOX或Val−Lys−PABC−DOX的前药可被cathepsin B高效切割,释放自由DOX,并在血浆中保持稳定。Arg虽被测试作为替代氨基酸,但表现不如Lys;而与Arg同构的瓜氨酸(Cit)在Val−Cit−PABC−DOX中则展现出良好的血浆稳定性。不同肽基对DOX释放速率存在差异,例如Z-Phe−Lys−PABC−DOX的释放速率比Z-Val−Cit−PABC−DOX快30倍,但在大鼠肝溶酶体中两者速率相同。比较来看,Val−Cit连接子在血浆稳定性优于Phe−Lys连接子,两者均显示出显著抗肿瘤活性。
目前,Val−Cit连接子是研究最为广泛的溶酶体可切割二肽连接子,已用于6种FDA批准药物中。另一类Val−Ala连接子也被证实为有效的蛋白酶可切割基团,并已用于批准药物loncastuximab tesirine (Table 2)。在cathepsin B体外实验中,Val−Ala的切割速率约为Val−Cit的一半,但其疏水性较低,且具有更高的药物−抗体比(DAR),并能减少ADC聚集。进一步比较显示,带有Val−Ala连接子的ADC抗肿瘤活性优于Val−Cit、Val−Lys与Val−Arg类似物。近期,Val−Gln连接子在细胞裂解液与cathepsin中切割速度更快,且含有Val−Gln的ADC展现出强效抗肿瘤活性(Table 2)。这些结果均表明,酶切型肽连接子是ADC药物中的关键组成。
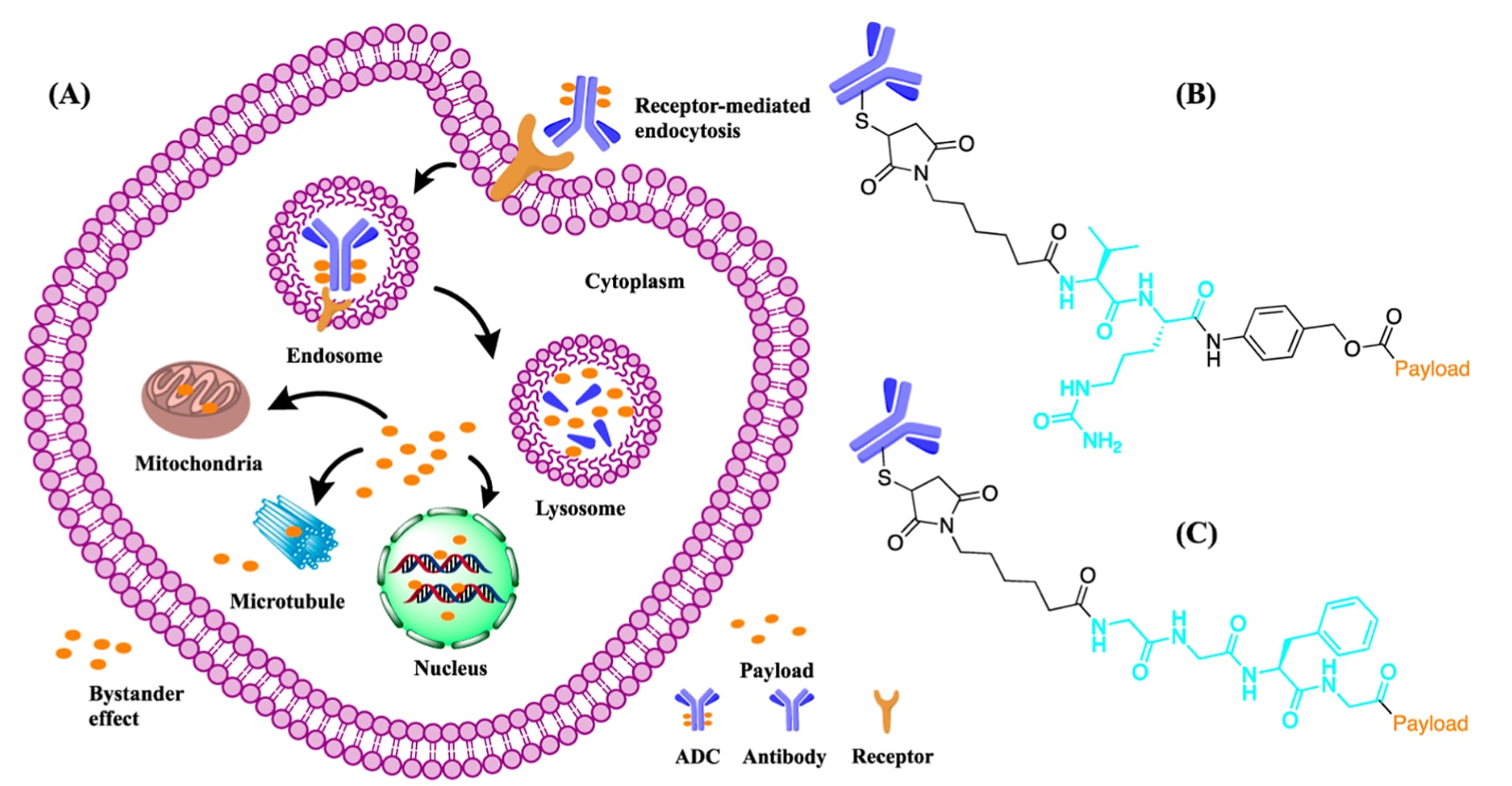
图2 (A) 传统ADC作用机制:ADC的抗体与癌细胞表面受体结合,形成ADC−抗原复合物,并通过受体介导的内吞作用进入细胞。随后,ADC从内体转运至溶酶体,在此降解释放载荷至细胞质中。游离载荷通过与微管、线粒体或细胞核内DNA相互作用来杀伤肿瘤细胞。一部分载荷还可扩散至邻近肿瘤细胞,产生旁观者效应。(B) Val−Cit二肽连接子的化学结构。(C) Gly−Gly−Phe−Gly四肽连接子的化学结构,均以青色标注。
2.2 Val−Cit与Val−Ala二肽连接子的缺陷
尽管Val−Cit或Val−Ala二肽连接子在人体血浆中稳定,且能在靶细胞溶酶体中高效释放药物,但仍存在一些局限:
- 在啮齿类动物中不稳定:因其易受啮齿类血浆中胞外丝氨酸水解酶carboxylesterase 1C(Ces1C)降解,从而导致药物过早释放,影响临床前安全性与疗效研究的准确性。
- 人源中性粒细胞弹性蛋白酶的影响:该酶可水解Val−Cit连接子,导致骨髓中分化中性粒细胞分泌的丝氨酸蛋白酶介导的胞外切割,从而引发中性粒细胞减少症。
- 疏水性诱导聚集:Val−Cit或其他二肽连接子通常需要PABC间隔子,而其疏水性与Val−Cit自身疏水性叠加,容易诱导ADC聚集。研究发现,亲水性葡萄糖醛酸连接子可稳定形成单体ADC,而Val−Cit连接子则易导致ADC聚集。此外,连接子与载荷增加抗体表面疏水性,进一步促进聚集并增强非特异性蛋白相互作用,进而提高脱靶毒性风险。
虽然通过降低DAR可缓解疏水性带来的负面影响,但对于低表达靶点或中等效力载荷而言,该方法并不适用。鉴于Val−Cit与Val−Ala连接子的局限性,研究者已开发出多种新型cathepsin可切割肽连接子来克服这些问题(Table 2)。
表1 | 全球已上市的ADC及其特征
2.3 新型可被Cathepsin切割的肽连接子
带有Glu−Val−Cit连接子的ADC表现出良好的酶切药物释放效果,并且在小鼠体内几乎不发生过早切割。另一种采用喜树碱衍生物作为载荷、通过新型亲水性cathepsin可切割的Val−Lys−Gly三肽连接子偶联的ADC,在实体瘤与血液肿瘤的临床前研究中均展现出广谱治疗活性。Glu−Gly−Cit连接子在小鼠和灵长类模型中保持长期稳定性,并能抵抗中性粒细胞蛋白酶介导的降解,含有该连接子的ADC在乳腺癌治疗中表现出更佳的疗效与安全性。使用Val−Ala−Gly三肽连接子的ADC抗肿瘤活性优于已批准的Kadcyla。该连接子虽不含PABC自裂解基团,但能在靶细胞溶酶体内切割释放药物。
Gly−Gly−Phe−Gly四肽连接子最早在DOX与羧甲基普鲁兰结合物中被发现,表现出优于无连接子的偶联物的抗肿瘤效果。后续研究证明,该四肽连接子的药物释放依赖于肿瘤中cathepsin的表达。新型ADC DS-8201a (T-Dxd, trastuzumab deruxtecan),采用此类连接子,对高或低HER2表达的T-DM1耐药乳腺癌患者均有效。另一种TROP2靶向ADC Datroway (datopotamab deruxtecan) 也含有该四肽连接子,已被批准用于治疗不可切除或转移性乳腺癌患者。这些结果表明,三肽或四肽连接子是一种提升ADC稳定性与安全性的有效策略。
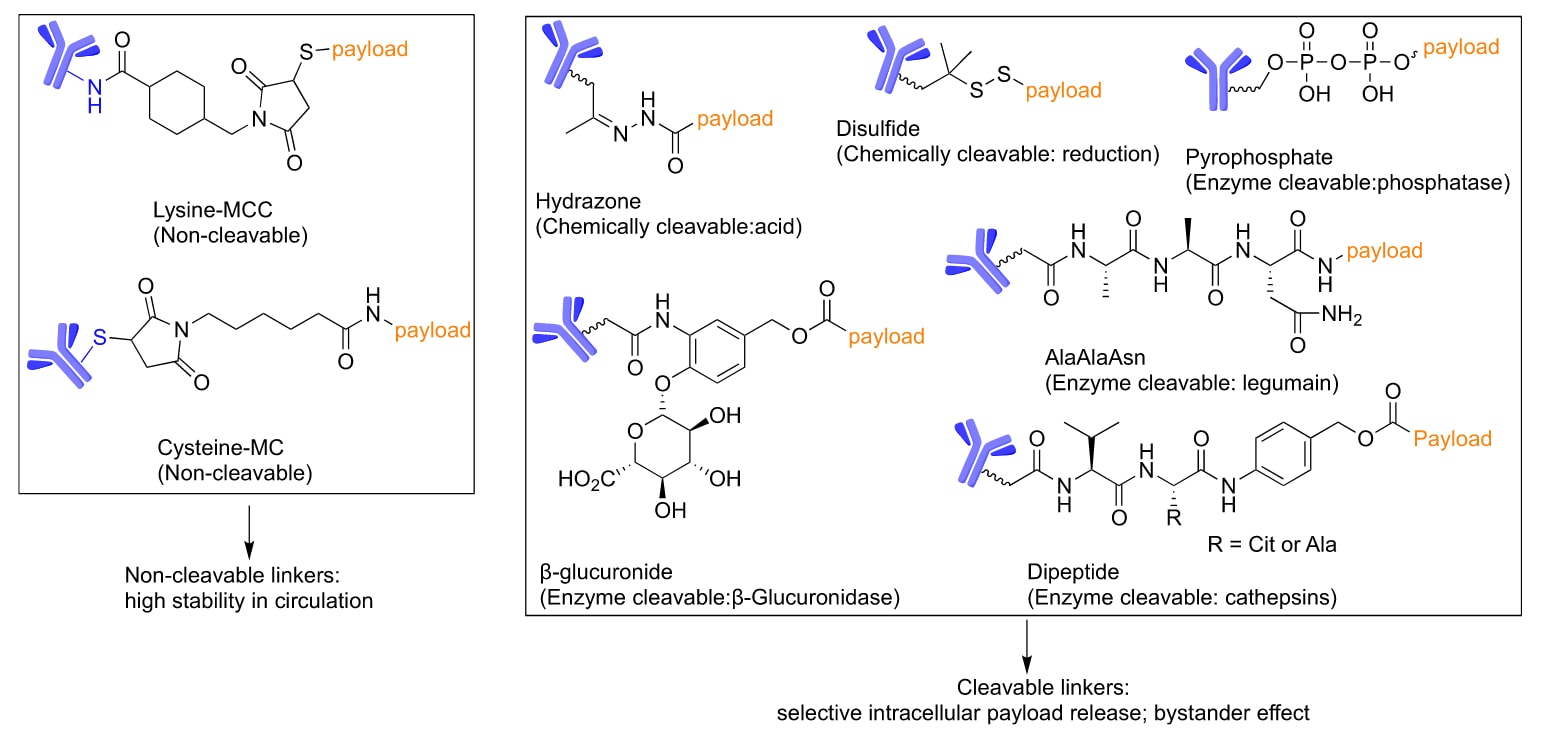
图3 | ADC连接子的化学结构与特征。 不可切割连接子主要包括MCC(马来酰亚甲基环己烷-1-羧酸酯)和MC(马来酰己酰)硫醚连接子。可切割连接子则可被酸性环境(如内体与溶酶体)、还原性分子(如谷胱甘肽)或酶切割。
表2 | 含肽连接子的酶激活型ADC
2.4 利用其他酶切型肽连接子
目前多数已上市ADC采用cathepsin可切割肽连接子。然而,研究发现这些连接子也可能被外源酶(如Ces1C和中性粒细胞弹性蛋白酶)切割,从而导致脱靶毒性。人类基因组中已鉴定出600余种蛋白酶,其中许多与肿瘤增殖、侵袭和转移密切相关。因此,研究者开发了基于**其他酶类(如legumain、纤溶酶PLM和成纤维细胞活化蛋白FAP)**的肽连接子,以减少非特异性药物释放(Table 2)。
- Legumain连接子:Legumain是一种半胱氨酸内肽酶,在大多数人类实体瘤中过表达,并与预后不良和晚期病程相关。其特异性要求底物在P1位带有Asn。基于Ala−Ala−Asn−Leu四肽或Ala−Ala−Asn三肽的legumain激活前药已被设计,并在多种肿瘤中展现有效抗癌活性。将Val−Cit连接子与Ala−Ala−Asn连接子结合的ADC显示出强效抗肿瘤活性。含有Asn−Asn二肽连接子的MMAE-ADC具有与Val−Cit ADC相当的强效和选择性,并且对中性粒细胞弹性蛋白酶完全稳定,在小鼠和人血清中也表现出优异稳定性,凸显了legumain切割型连接子的巨大潜力。
- 纤溶酶(PLM)连接子:PLM在肿瘤侵袭与转移中发挥重要作用。研究合成并测试了一系列三肽酰胺作为底物,其中Ala−Ala−Lys三肽是PLM的特异且稳定底物。基于D-Val−Leu−Lys的前药对癌细胞的选择性比游离药物提高5倍。进一步开发的DOX和PTX前药(如D-ala−Phe−Lys−PABC−DOX、D-val−Leu−Lys−PABC−PTX)展现出优异抗癌活性和更快的PLM激活速率。一种带有Val−Leu−Lys连接子与MMAE载荷的ADC在抑制肿瘤生长方面显著优于对照组。
- FAP连接子:FAP是一种丝氨酸蛋白酶家族的细胞表面糖蛋白,由癌相关成纤维细胞在90%以上的上皮癌中高表达,而在正常成纤维细胞及其他正常组织中几乎不表达,因此是极具潜力的治疗靶点。FAP的底物特征为严格的Gly−Pro切割特异性。含有Gly−Pro连接子的ADC可在肿瘤部位选择性释放药物,在临床前模型中展现出良好抗肿瘤效果。此外,FAP切割的Gly−Pro二肽连接子在多种SMDCs中也表现出快速而特异的肿瘤微环境切割。

图4 | 代表性ADC连接子的化学结构 ,含有亲水性基团的修饰:PEG(a−c)、环糊精(d)与pSAR(e),青色背景部分为高亮。
3 提升ADC治疗效果的连接子修饰策略
在ADC设计中,理想的连接子应具备以下特性:良好的溶解性、在细胞外循环中的稳定性以及在溶酶体内快速释放载荷。连接子的长度、极性与溶解性不仅影响ADC的稳定性与靶向结合能力,还直接决定载荷释放效率与整体治疗效果。近年来,研究者提出了多种策略来优化连接子的性质,从而提升ADC的治疗效果。
3.1 在连接子中引入亲水基团
ADC中肽连接子的一个普遍特点是疏水性较强,当连接子与载荷均为疏水性时,往往限制了ADC的药物−抗体比(DAR)。在靶抗原密度较低时,高DAR偶联物可增强治疗效果,但同时也会加速血浆清除,降低体内疗效。因此,引入亲水性基团以平衡疏水性载荷成为一条重要途径。
聚乙二醇(PEG)是应用最广泛的亲水聚合物,因其水溶性高、免疫原性低且内在毒性小,被广泛用于医学领域。已获批的ADC sacituzumab govitecan (Trodelvy) 与 loncastuximab tesirine (Zynlonta) 都引入了PEG基团(Figure 4a),而且PEG在ADC试剂中的应用已被深入研究。带有磺酸基或PEG的亲水连接子能在高DAR条件下偶联疏水性载荷,而不会引起聚集或亲和力下降,这优于传统使用的疏水性SMCC类连接子。PEG化修饰不仅提高了ADC的亲水性和DAR,还显著改善了其体内药代动力学特性。
在Val−Lys二肽连接子中,PEG与赖氨酸游离氨基偶联可提升ADC的稳定性、亲水性与治疗效果,相关研究已在紫杉醇基ADC与MMAE基ADC中得到验证(Figure 4b)。进一步研究发现,PEG的臂数、每条臂上的氧化乙烯单元数以及切割点与PEG分支间的距离均会影响酶介导的载荷释放效率。新型含有双PEG12分支的Val−Ala−Gly连接子在卵巢癌小鼠模型中表现优于Kadcyla,展现出更高治疗效果(Figure 4c)。另一种含分支PEG修饰的酶切肽连接子的ADC OBI-992,在多种癌症模型中展现出卓越的稳定性与抗肿瘤活性。
除了PEG外,环糊精(cyclodextrin, CD) 或冠醚等亲水性大环结构也能通过谷氨酸分支点引入ADC中,在部分研究中其疗效优于已上市的brentuximab vedotin(Figure 4d)。此外,聚肌氨酸(polysarcosine, pSAR) 也被用作疏水性屏蔽基团,开发了高DAR、均一且对β-葡萄糖醛酸酶敏感的ADC。在此平台上,基于exatecan的ADC在异种移植模型中表现出强效抗肿瘤活性,甚至优于FDA批准的DS-8201a(Figure 4e)。
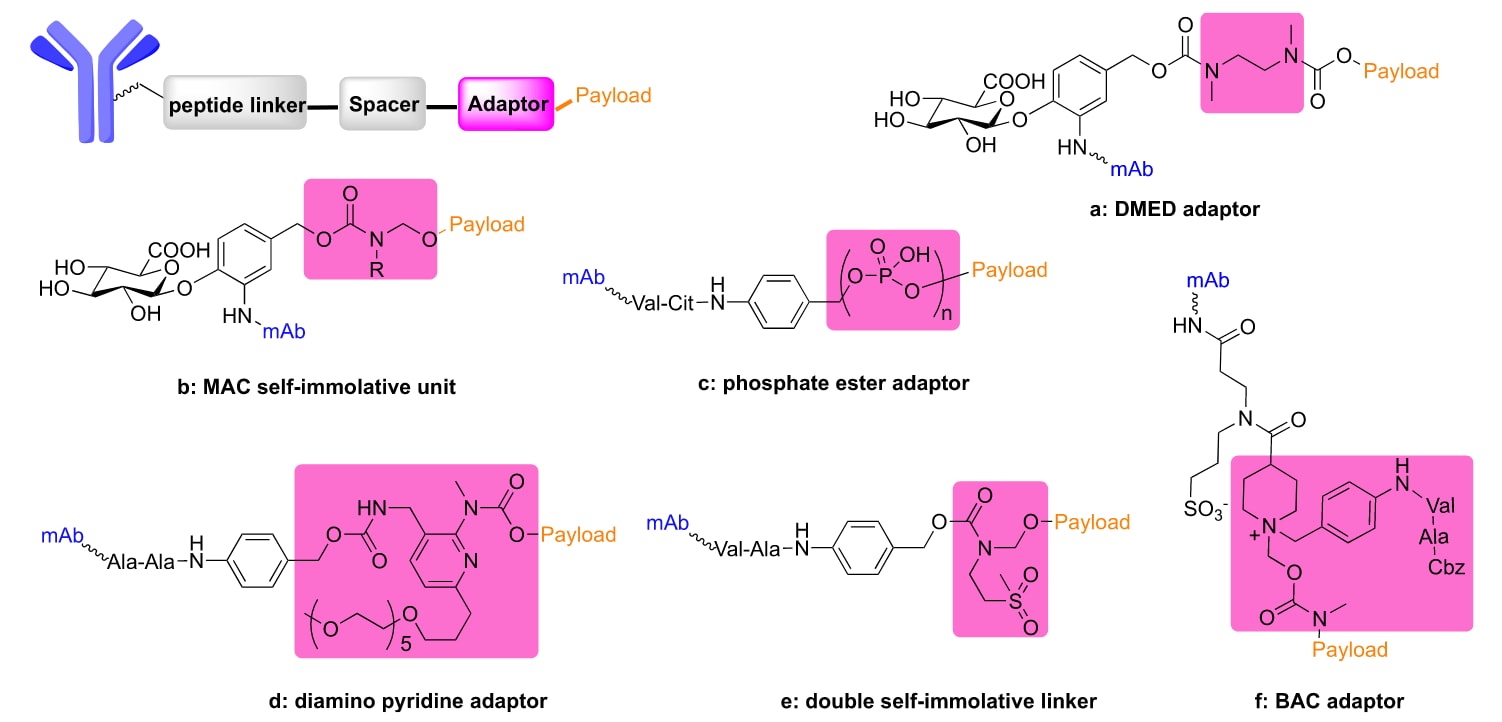
图5 | 连接子修饰中的新型适配体的化学结构(a−f), 粉色背景部分为高亮。
3. 2 设计ADC连接子的新型间隔子或适配体
自裂解(self-immolative, SI)间隔子是一类共价组装结构,能够触发级联解离反应,从而释放载荷。SI间隔子的裂解依赖于邻位或对位的离域效应。目前,大多数上市的ADC采用Val−Cit或Val−Ala二肽,其后接SI间隔子PABC,以释放结构不变的药物。传统的 MC−Val−Cit−Ala−PABC 连接子技术要求载荷必须具备氮原子连接位点以形成氨基甲酸酯。虽然可以通过有机合成引入氨基,但生成的药物类似物可能对ADC的药理性质产生不利影响。碳酸酯或酯类连接策略虽可用于ADC,但其血浆稳定性有限,半衰期较短。因此,亟需开发将含羟基的载荷与抗体偶联的新方法。
一种解决方案是在SI间隔子与载荷之间引入适配体(Figure 5)。早在2001年,研究者将PABC与N,N′-二甲基乙二胺(DMED)偶联,设计了纤溶酶或β-葡萄糖醛酸苷酶激活的紫杉醇前药。这类新型前药相比传统间隔子的前药具有更快的激活速度和更低的细胞毒性。为了拓展β-葡萄糖醛酸连接子在含酚类药物中的应用,研究者将DMED适配体引入连接子,所得ADC表现出高效的药物释放和抗肿瘤活性(Figure 5a)。进一步研究表明,吡咯烷适配体或含叔胺的吡咯烷适配体能显著影响SI间隔子的环化效率,从而调控药物释放速率和抗癌活性,尤其适用于含伯醇类载荷(Figure 5b)。
另一种方法是使用亚甲氧基(MAC)适配体。MAC单元与可酶切连接子结合后,能在ADC中实现对含羟基载荷的高稳定性和高效释放。针对疏水性醇类载荷,研究者开发了磷酸酯适配体。磷酸酯桥接cathepsin B敏感连接子,可使载荷通过羟基结合,并在溶酶体环境中快速释放,且在血浆中表现出优异的稳定性(Figure 5c)。
此外,研究者还开发了无痕SI连接子,其基于邻位或对位羟基苄胺,能够在水溶环境下保持稳定,并维持蛋白等电点。所得偶联物保留了氨基上的正电荷,减少了因等电点变化导致的蛋白变性和聚集(Figure 5d)。另一种二氨基吡啶适配体可将PABC稳定地连接到布地奈德的羟基上,PEG修饰后显著降低ADC聚集性,提高了血浆稳定性和抗肿瘤活性。
近期,还开发了一种双SI连接子策略,结合双胺自环化与PABC双重间隔子,能够高效偶联含羟基的载荷。引入2-氨基乙基甲基砜增加了空间位阻,进一步提升了ADC的稳定性(Figure 5e)。另一种新型苄基α-铵基氨基甲酸酯(BAC)连接子可通过1,6-和1,2-连续消除反应选择性地在肿瘤中释放载荷,且细胞毒性优于传统的PAB−Val−Cit类ADC(Figure 5f)。
尽管SI的1,6-消除反应效率很高,但其适用范围局限于胺(氨基甲酸酯)、醇或酚(碳酸酯)等酸性官能团。为突破这一限制,研究者设计了基于醌亚甲基的新型SI间隔子,能够在还原刺激下直接连接并释放胺、酚、硫醇、磺酰胺和酰胺。同时,开发了一种三功能SI间隔子,只有在蛋白酶与磷酸酶双重作用下才触发药物快速释放。最近研究还发现,带有简单酯键的ADC(连接在二肽连接子与含羟基载荷之间)在体外表现出优异活性,在小鼠血清中稳定,并能在人肝溶酶体中快速切割。但仍需进一步体内研究以评估其与经典ADC的对比效果。
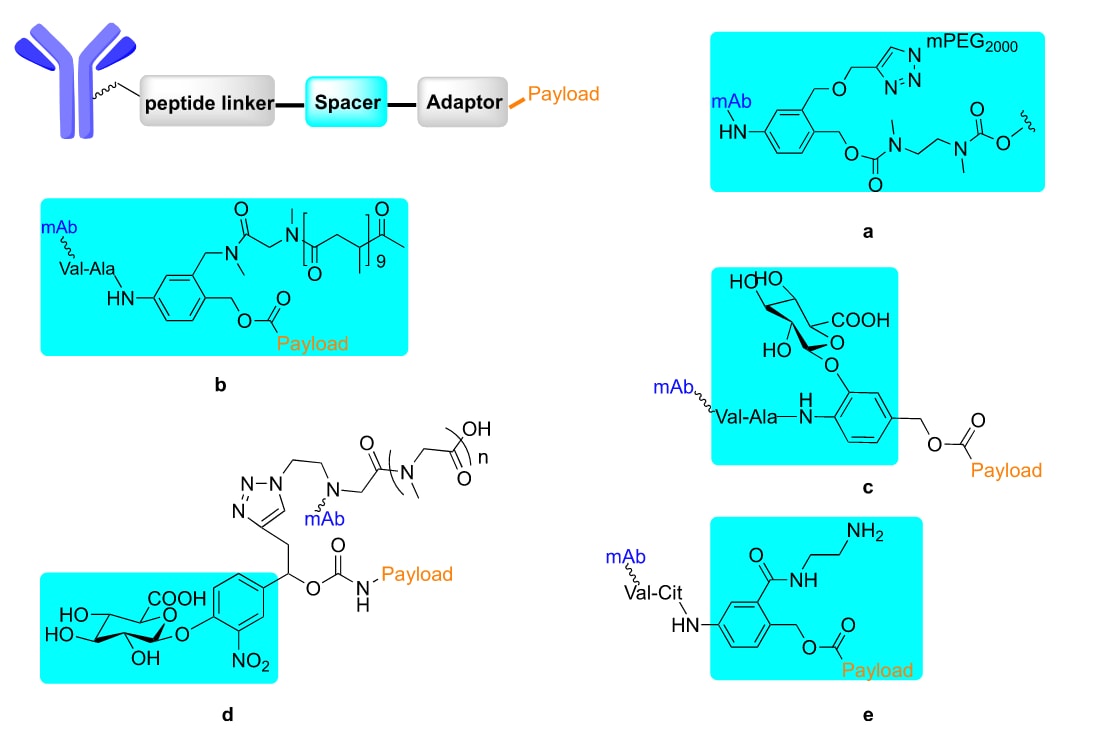
图6 | ADC连接子中PABC修饰的化学结构(a−e)。 苯环上的自裂解型PABC修饰以青色标注。
3.3 ADC连接子中PAB的修饰
对 PABC间隔子区域 的修饰也会显著影响ADC的血清稳定性、亲水性以及聚集性(Figure 6)。例如:
-
mPEG2000基团 引入PABC的苯环(Figure 6a),可显著提高前药纳米颗粒的水溶性。
-
在PABC间隔子中加入 pSAR基团,使ADC的聚集率降低至0.64%,并实现了高达 DAR=8 的药物偶联比(Figure 6b)。
-
葡萄糖醛酸(glucuronide) 和 α-L-艾杜糖醛酸(α-L-iduronide) 触发策略,作为高效的SI连接子,能选择性地将载荷递送至肿瘤部位。
-
双切割连接子 策略:在PABC苯环上引入葡萄糖醛酸基团(Figure 6c),通过两步连续的酶切反应释放载荷,增强了ADC在循环中的稳定性。
-
双酶切3-O-硫酸-β-半乳糖连接子 能够依次被溶酶体的 芳基硫酸酯酶A 和 β-半乳糖苷酶 切割,从而在靶细胞中释放载荷。
-
将 葡萄糖醛酸SI基团 与 pSAR亲水性遮蔽基团 结合使用,可提升药物负载量,改善理化性质、药代动力学特性,并在体内增强抗肿瘤疗效(Figure 6d)。93
-
为了提高 Val−Cit−PABC连接子体系 在小鼠血浆中对 Ces1C酯酶 的稳定性,研究者尝试了多种PABC修饰方式,包括:
-
用噻唑(thiazole)等杂环取代苯环
-
在苯环上进行不同的取代修饰
结果表明,在苯环中引入 酰胺基 效果最佳(Figure 6e)。
-
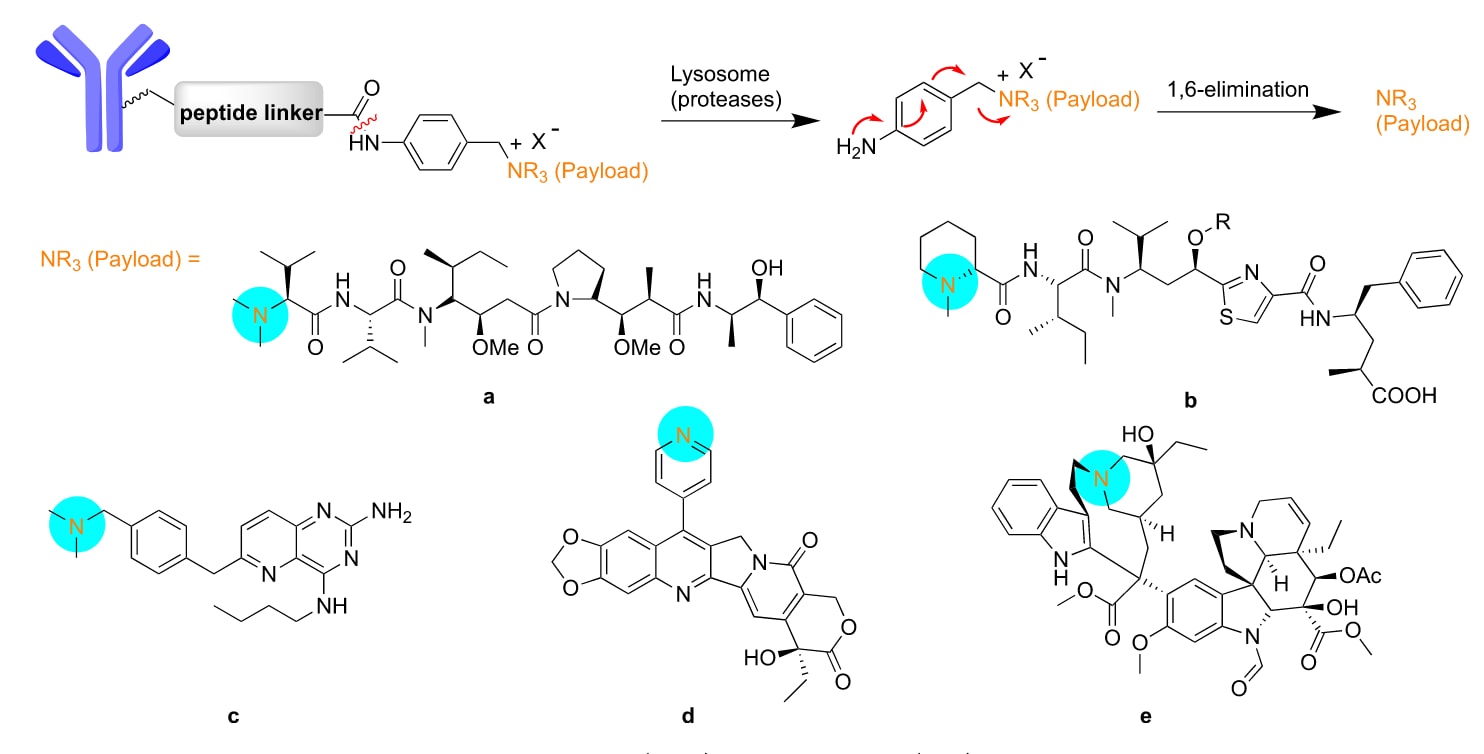
图7 | ADC连接子中季铵盐(QAS)的化学结构(a−e)。 如图上方所示,酶切断酰胺后,p-氨基苄基QAS自裂解,最终无痕释放出叔胺或杂芳胺与载荷。叔胺以青色标注。
3.4 季铵盐连接子
叔胺和杂芳基胺在天然产物和合成小分子药物中都很常见。一种新型的连接策略被设计出来,即通过对氨基苯基季铵盐(QAS)连接叔胺或杂芳基胺。所得的季铵盐(QAS)连接子在抗体偶联药物(ADCs)中能引入带电的官能团,从而提高水溶性、减少聚集并改善偶联物的性质。QAS 连接子已成功应用于蛋白酶可切割型和还原可切割型的 ADCs 中,这些偶联物相较于氨基甲酸酯连接的偶联物表现出更高的稳定性和活性(图7a)。
由于微管抑制活性,Tubulysin 是有前景的抗癌细胞毒剂。通过 QAS 连接子将抗体与 Tubulysin 偶联,设计出了新型 ADCs。当 R 基团由乙酸酯改为丙氧基时,其稳定性和抗癌疗效均得到改善(图7b)。
利用 QAS 连接策略,将抗 PD-L1 抗体与双功能免疫调节剂偶联,构建了一种免疫调节型 ADC,用于癌症免疫治疗(图7c)。该 ADC 的疗效优于抗 PD-L1 抗体与免疫调节剂的组合治疗。
在喜树碱第 7 位引入吡啶基显著增强了这些衍生物的细胞毒活性。利用这些喜树碱衍生物与 QAS 连接子构建的新型 ADCs 显示出优异的血浆稳定性(图7d)。进一步在该连接子上修饰甲磺酰乙胺,可提高 ADC 在体外的细胞活性。
长春新碱(Vinblastine)和长春花碱(Vincristine)是被批准用于多种癌症的化疗药物。利用含有 QAS 的可切割连接子对长春花碱进行 N-烷基化,得到的曲妥珠单抗−长春花碱偶联物在体外对 HER2 阳性细胞表现出亚纳摩尔级效力,远低于游离长春花碱,且与已上市的 ADC 相当(图7e)。
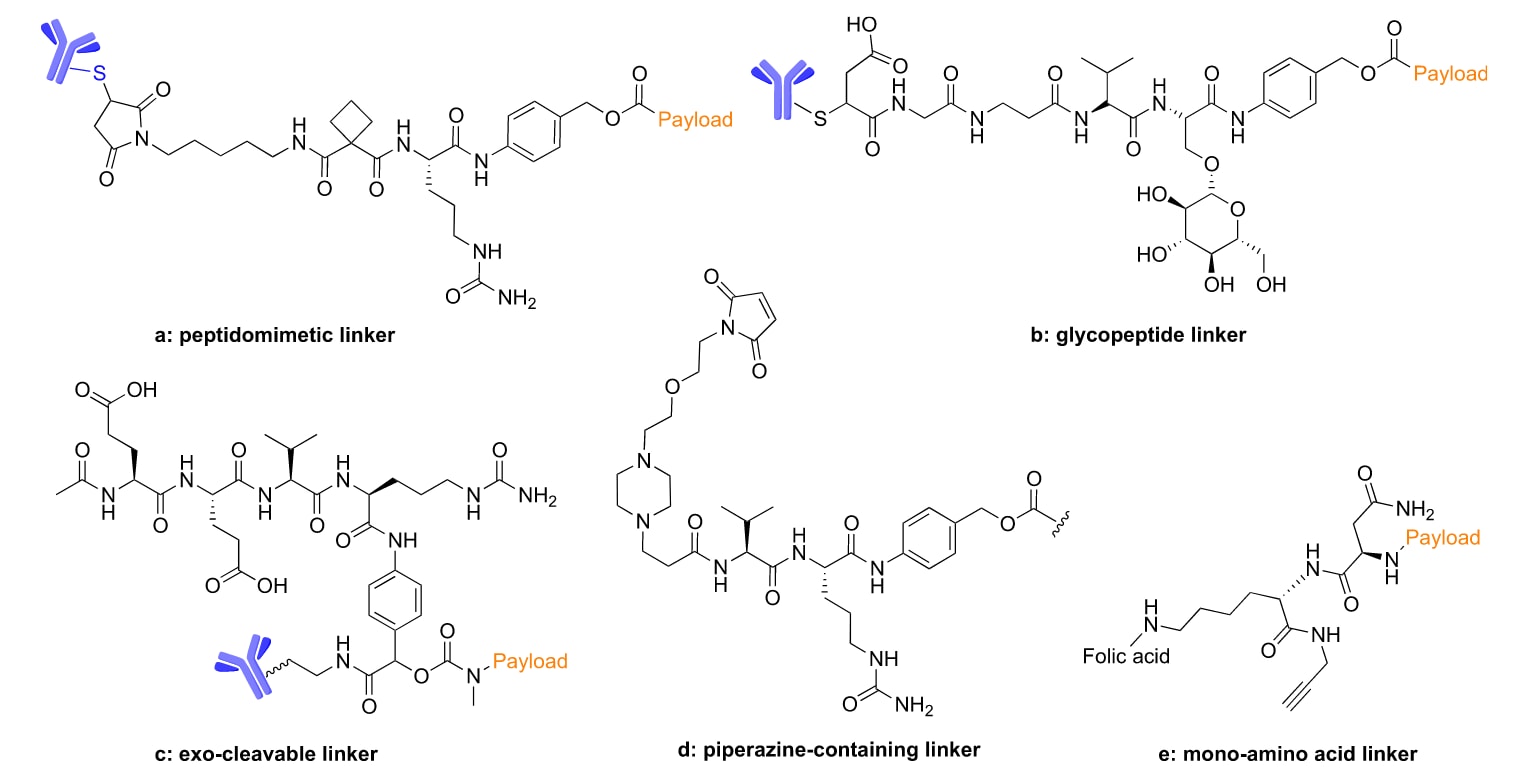
图8 新型连接子的化学结构(a−c),这些连接子可提升ADC的血清稳定性与治疗效果;(d) 前药;(e) SMDCs。
3.5 前药、SMDCs 或 ADCs 中的新型肽类连接子
为了进一步提高对组织蛋白酶 B(cathepsin B)的选择性,研究者基于该酶的结构,合理设计了一种含有肽拟态环丁烷-1,1-二羧酰胺的连接子(图8a)。采用这种连接子的 ADC 在小鼠肿瘤模型中表现出更好的组织蛋白酶 B 选择性,并且与带有 Val−Cit 连接子的偶联物相比,具有相当的稳定性。当其与吡咯并二氮杂䓬二聚体(PBD)偶联时,表现出比 Val−Cit−PBD ADC 更强的抑瘤作用。这一结果表明,具有增强蛋白酶特异性的肽拟态连接子在新型 ADC 构建中的应用前景。
另一种新型糖肽连接子被开发出来,利用该稳定化糖肽连接子的 ADC 显示出雄酰亚胺(maleimide)的稳定性增强、对血清酶切割的抗性更强,并且疗效优于 Val−Cit 曲妥珠单抗 ADC(图8b)。
外切可裂解(exocleavable)连接子将可裂解肽连接子重新定位到 PABC 结构单元的外部位置,从而微妙地降低了药物过早释放的风险(图8c)【133】。使用这种连接子的 ADC 表现出更高的稳定性和治疗效果。
除了 ADC 之外,一些新型连接子也被开发用于前药和小分子药物偶联物(SMDCs)。一项最新研究发现,在连接子中引入哌嗪基团与参考的含 PEG 连接子相比,显著提高了前药的水溶性(图8d)【134】。这种连接子的前药表现出很高的水解稳定性、优异的白蛋白结合能力和强大的肿瘤富集效果。
另一项引人注目的研究表明,带有单氨基酸天冬酰胺(Asn)连接子的 SMDC 在效力方面有显著提升,并且对内涵体中组织蛋白酶 B 的切割反应快速(图8e)【135】。
4 结论与展望
ADC 在治疗血液恶性肿瘤和实体瘤方面已取得巨大成功。可切割肽连接子在临床肿瘤学中展现出良好的安全性。然而,肽连接子过早释放载荷仍存在风险,可能导致系统性毒性并降低药物递送效率。因此,如何在循环中保持稳定性,同时又能在肿瘤部位被激活,是未来肽连接子开发中的关键挑战。
最近,研究人员开发了一种缺电子的磺酰胺类可切割连接子,结果表明调节芳基磺酰胺的吸电子特性对于平衡 ADC 的稳定性和药物释放至关重要。ADC 的聚集是主要关注点之一,因为其可能增加非靶向细胞毒性并削弱注射药物的安全性和有效性。环糊精(CDs)能够溶解小分子药物并稳定蛋白质;最新研究表明,合理的 CD 制剂可以改善 ADC 的物理稳定性,并减少不同载荷和连接子所导致的聚集。
连接子在 ADC 的稳定性、水溶性、载荷释放速率以及非靶向毒性中发挥着关键作用。连接子化学的发展使得能够构建出更安全、更高效的 ADC,用于治疗多种肿瘤。尽管组织蛋白酶 B 并非 cathepsin B 可切割 ADC 所必需,但关于连接子切割机制的研究仍然有限,而这对于肽连接子的设计却十分重要。对抗体内吞及药物释放的研究已成为理解 ADC 作用机制并指导其设计的关键。
ADC 内吞和药物释放的关键因素可以通过一种名为 TORCH(Turn On after Release by CatHepsins)的新型分子来评估。TORCH 含有一个被荧光淬灭剂抑制的系统,该系统通过组织蛋白酶 B 可切割的二肽连接子分离。开发能够实时监测 ADC 亚细胞水平载荷释放的方法,将有助于肽连接子的合理设计。
新型肽连接子有望在降低毒性和耐药性的同时增强 ADC 的治疗效果,并最终为患者带来临床获益。