BioDesign Research 2022 | 用于结合蛋白靶标的蛋白片段与肽的设计

Gupta, S.; Azadvari, N.; Hosseinzadeh, P. Design of Protein Segments and Peptides for Binding to Protein Targets. BioDesign Research 2022. https://doi.org/10.34133/2022/9783197.
0 摘要
近年来,精确预测蛋白质结构与设计新型功能蛋白的方法不断涌现。功能性蛋白片段与肽的设计虽在整个蛋白设计领域中占据较小但独特的地位,却具有重要意义。由于这些肽分子体积更小,计算方法能够进行更为全面的搜索与优化,但与此同时,它们的结构灵活性更高、可用数据相对稀缺,并且常常包含非天然氨基酸等非典型结构单元,这些因素都给设计带来了额外的挑战。该文章综述了当前在用于靶标结合的蛋白片段与肽设计方面的最新进展,并探讨了该领域所面临的主要问题与未来的发展方向。
肽(即由短链氨基酸构成的分子,一般少于50个残基)在细胞功能中发挥着关键作用(图1,表1)。许多肽充当激素,在体内传递信号,例如代谢激素胰岛素与神经肽催产素均属于典型的肽类激素。部分蛋白片段(下文也称为肽)则负责调控细胞信号传导通路,如信号肽将蛋白引导至正确的亚细胞定位,而富脯氨酸肽则通过与SH3结构域相互作用参与多种信号途径。一些肽还具有防御功能,抗菌肽(AMPs)、多种抗生素(如革兰阳性菌素S与lantibiotics)以及部分毒素均属此类。肽在功能上的多样性表明,通过设计肽分子可实现从调节细胞信号到开发新型抗生素等多种应用。

图1 | 肽可呈现多种结构形式。 图中展示了若干天然肽的结构。棒状线条表示交联键;洋红色部分标示非天然氨基酸;绿色残基为D-型氨基酸;球体表示侧链C-β原子的空间位置;虚线则代表氢键。
1 引言
除了天然功能外,肽也被广泛用作治疗分子,是抗体与小分子药物的有益补充。与抗体类似,肽能够以高亲和力和高选择性结合于平坦的蛋白表面;与小分子类似,它们可以穿透细胞膜以靶向胞内蛋白。因此,肽提供了一种独特机会,可用于靶向目前被认为“难以药物化”的疾病相关蛋白。而当前获得此类治疗肽的方法往往依赖文库筛选,因此,能够定向设计具有期望特性的肽以引导文库构建成为研究热点。
尽管肽在天然生理与药物开发中均具有重要地位,功能性肽的设计依旧极具挑战。除了蛋白设计领域共有的困难外,肽设计还面临自身独特的限制。首先,在结构建模方面存在实际瓶颈。肽序列中丰富的非天然氨基酸(ncAAs)需要在力场参数中进行额外定义,这些氨基酸赋予肽独特的构象特征,而这些特征在现有蛋白结构数据库中并不存在。许多肽还包含交联基团或环化结构,这会对主链构象施加限制,使得键长与键角偏离天然蛋白中的平衡值,例如脯氨酸环的异常扭曲或非平面肽键等。这意味着,基于蛋白结构所得的启发式知识往往不足以准确建模肽的三维结构,而基于蛋白数据库训练的学习模型也难以直接推广到肽分子。
另一个困难在于,肽的结构与功能关系模糊。与结构稳定的功能蛋白不同,许多功能性肽具有内在无序性或多构象状态。这种构象可塑性在设计中造成两方面问题:其一,增加了设计的复杂度;其二,由于这些多构象状态难以实验验证,可用于模型学习的实例显著少于稳定蛋白。此外,由于尺寸较小,肽常缺乏蛋白设计中的基本单元——二级结构。
设计能结合蛋白表面的功能性肽还需应对正确建模结合构象的挑战。此类构象的采样通常依赖分子对接或分子动力学模拟,但这些方法往往计算成本高且精度有限,原因包括采样与打分不足以及界面水分子的建模困难。众所周知,许多肽与靶标蛋白之间通过水介导氢键相互作用。然而,尽管当前在水分子计算建模方面已有进展,精确描述界面水网络仍是计算设计中的难题。
尽管如此,研究者已成功设计出多种功能肽,包括可特异结合靶蛋白的肽(综述的主题)、催化肽以及基于肽的高阶自组装结构。总体而言,当前的设计策略可分为三大类(图2):(a) 结构引导型设计,(b) 从头设计,以及(c) 基于学习的设计方法。需要指出的是,这些方法之间的界限并非绝对,而是相互交织。以下部分将重点介绍以靶标结合为主要应用的肽设计策略及其代表性进展,以呈现该领域的整体发展图景。

表1 | 不同类型肽的分类说明、特征及其结构实例。 总结了多类肽的基本信息,包括其功能描述、主要结构特征以及具有代表性的结构实例,以展示肽分子在生物功能与构象多样性方面的广泛差异。
2 基于结构引导的功能肽设计
在功能性肽的设计中,结构引导型设计是迄今最成功的策略之一。该方法通常以一个**已知具备目标功能的天然片段(一般≥4个残基)或结构基序(motif)**为起点,在此基础上进行改造优化。这类改造往往较小,保留了原有序列和空间构象。例如,整合素结合基序RGD环便是这类代表性实例之一。此类受天然肽基序启发的方法被统称为结构引导型设计。
这类基序通常从蛋白质数据库(PDB)中提取的天然蛋白结构中获得。然而,基序的识别与提取过程较为耗时,计算方法可显著加速这一过程。例如,Tsai等人构建了一个名为TP-DB(therapeutic peptide design database)的数据库,使研究者能够根据特定序列模式搜索匹配的螺旋基序。该数据库的模式特异性搜索引擎建立在从PDB中提取的约170万个螺旋结构的特征基础之上,这些被识别出的螺旋基序可作为肽设计的初始模板。
在另一项研究中,Alam等人开发并使用了一种名为FlexPepBind的算法,用于筛选组蛋白去乙酰化酶8(HDAC8)的潜在底物。FlexPepBind基于肽–蛋白复合物的结构模型计算肽与受体的结合特异性。研究者通过已知亲和力的肽集对其结果进行校准后,将该算法应用于人类蛋白组中具有乙酰化位点的所有肽段,从而成功发现了HDAC8的新底物。实验验证表明,HDAC8对26条候选肽中的20条表现出反应性,验证了算法预测的有效性。
在确定功能基序后,研究者通常通过以下几种方式进一步优化其性能(图3):
(a) 突变(mutagenesis),
(b) 交联(crosslinking),
(c) 支架化(scaffolding)。
值得注意的是,虽然基序在结合中起到关键作用,但其原始蛋白中的结合亲和力往往更高,因为在天然蛋白中,除基序外的邻近残基也对结合起到重要贡献。因此,为获得能与天然配体竞争的高亲和力肽,往往需要多种策略结合使用。例如,经过支架化的基序可进一步通过高通量突变文库实验优化,从而筛选出具有更强结合能力的肽分子。这种结合结构指导与实验筛选的混合设计框架,构成了当前功能肽设计的核心路径之一。
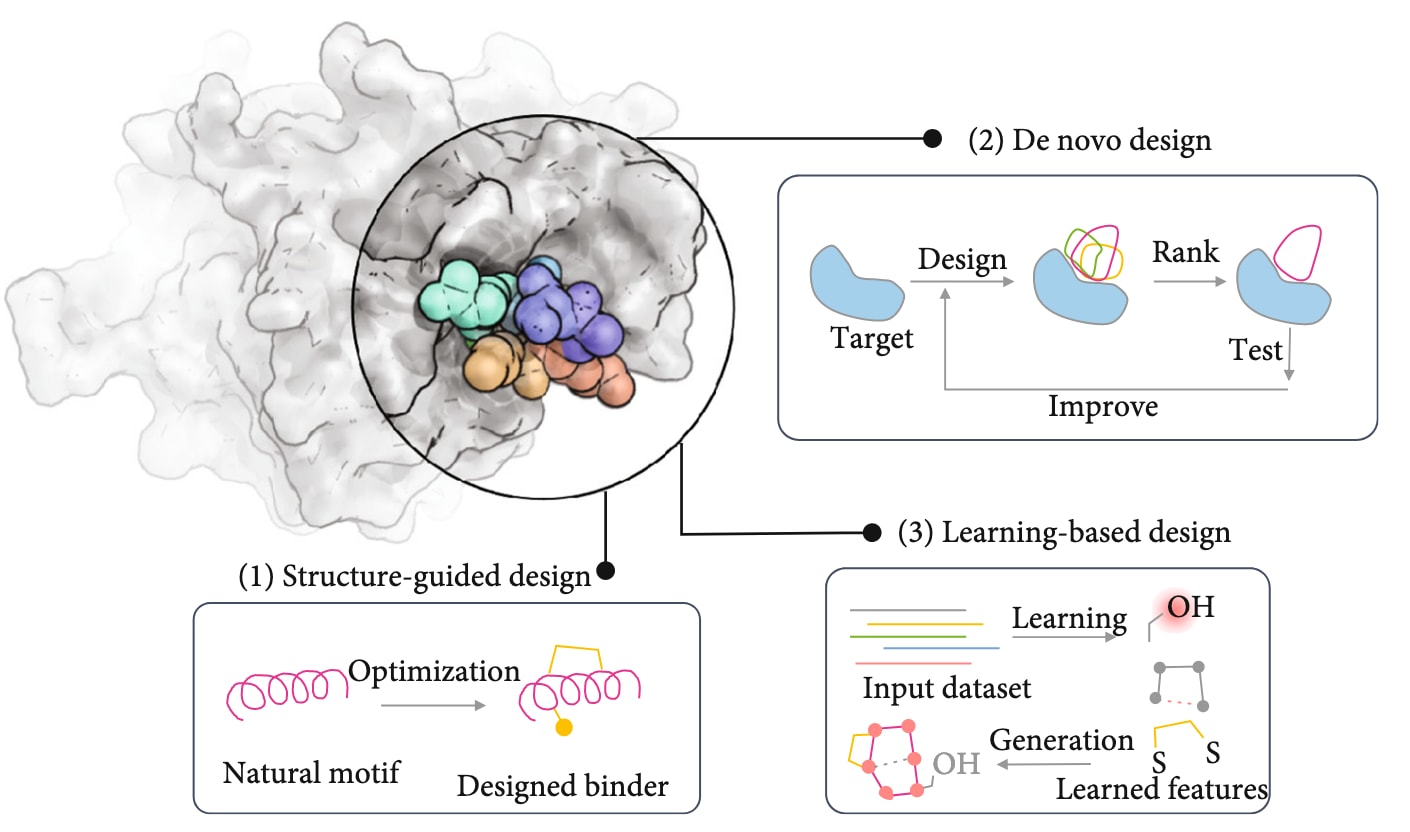
图2 | 用于设计肽类结合物的不同方法示意图。 为了获得能够结合目标表面的肽分子(图左上圆圈中球体所示,结构来自PDB编号:6WSJ),研究者通常采用三种主要策略:(1) 结构引导型设计,以天然存在的结构基序为起点构建结合肽;(2) 从头设计(de novo design),通过完全从零生成大量潜在肽分子并进行评分筛选,以确定最优候选;(3) 基于学习的设计方法,利用机器学习与深度学习技术,从现有实验与结构数据中学习规律,预测并生成高亲和力的肽结合物。
2.1 基于基序突变的结合肽设计
天然的蛋白–蛋白相互作用是在进化过程中形成以执行特定功能的,但这种功能并不总是与高结合亲和力直接相关。事实上,在许多生物过程中,低亲和力或瞬时结合事件反而是必要的。因此,通过在结合界面处对肽序列进行突变,可以增强已知肽–蛋白相互作用的结合强度(见表2)。
突变设计通常包括两个步骤:序列改动与效果评估。序列改动可以是完全随机的,也可以基于界面的生化知识进行引导,例如提高肽与靶标表面的形状互补性或增加氢键相互作用。评估步骤的复杂度从简单的结构可视化、到计算打分、再到高通量实验筛选均有不同层级。
一个典型且成功的实例是针对BCL-2家族蛋白的螺旋肽设计。通过结合计算突变、理性突变与实验筛选,研究者获得了多个具有高选择性和高亲和力的抑制剂,如靶向Bcl-xL、Bfl-1和Mcl-1的肽分子。这些工作展示了基于螺旋基序优化的强大潜力。Mackenzie等人进一步指出,利用**蛋白三级结构基序(TERMs)**的统计信息及其关联能量模型(dTERMen)可用于预测已设计肽与BCL-2家族成员的结合能,从而加速优化过程。
为实现非天然氨基酸(ncAAs)在肽设计中的自动化引入,Andrews等人开发了REPLACE算法(REplacement with Partial Ligand Alternatives through Computational Enrichment)。该方法首先以肽–靶标复合物结构为起点,将肽中目标氨基酸逐一移除,并将不同的功能基团对接到其位置处,通过打分函数进行排序。得分最高的基团被选为新的突变位点。利用此方法,研究者获得了能抑制CDK2但带电性更低、膜通透性更高的新型肽分子。
此外,Pearce等人利用EvoDesign算法在已知结合基序的基础上进一步寻找改进序列。EvoDesign借助蛋白–蛋白界面的进化信息设计更优结合界面。其方法是利用iAlign算法将非冗余蛋白界面库与目标界面对齐,生成多序列比对(MSA),并结合结构支架的进化特征作为引导信息。最终,这些进化能量项被整合进主能量函数EvoEF中以优化肽序列。研究者随后结合改进版能量函数EvoEF2,成功设计出可阻断SARS-CoV-2刺突蛋白与人ACE2受体结合的抑制肽。
在另一项研究中,Rooklin等人提出针对未占据口袋区域进行突变能显著提升结合亲和力。为辅助突变位点选择,他们开发了AlphaSpace算法,一种基于片段的界面口袋识别与分析工具。该算法的核心概念是AlphaSphere,通过对蛋白结构进行空间镶嵌后生成球体,再经过筛选、聚类与排序得到潜在口袋。当某口袋位于蛋白–蛋白界面时,可计算该位点残基与口袋的体积互补性;若差异足以容纳一个甲基基团,则该残基被标记为潜在突变目标。
在最新的研究中,作者利用AlphaSpace识别了MDM2与MDMX主结合位点邻近的次级口袋。通过局部对接找到最适配的侧链后,将原始结合肽延伸至该新识别区域。结果显示,延伸后的肽结合亲和力提高约一个数量级;在进一步对螺旋进行约束并优化连接基团后,最终获得了对MDM2和MDMX的结合常数分别为4 ± 8 nM和50 ± 16 nM的高亲和力肽,相比原始约1 μM的亲和力显著提升。
综上所述,基于基序突变的结构引导型设计通过在天然结合模板上进行系统性改造,结合计算筛选与实验优化,已成为提升肽结合性能的核心策略之一。
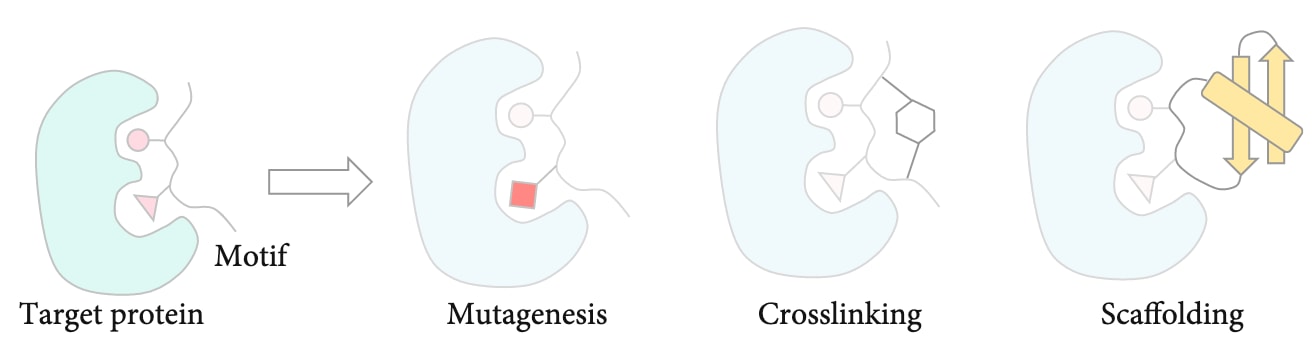
图3 | 结构引导型设计方法以天然存在的结构基序为起点。 该基序随后可通过突变(mutagenesis)、交联(crosslinking)或支架化(scaffolding)等方式进行稳定化与功能优化。通常需要多种策略的结合才能获得具有理想亲和力的肽分子。值得注意的是,在某些情况下,**从头设计(de novo design)**的肽也可在现有基序的基础上构建,并结合上述方法进行改进(详见第3节)。
表2 | 突变策略在肽结合物设计中的应用实例。 汇总了利用突变(mutagenesis)方法优化肽–蛋白相互作用的典型研究案例,展示了通过序列改造、界面优化或非天然氨基酸引入等手段提升肽的结合亲和力与选择性的多种实践途径。
2.2 使用交联剂稳定结构基序
肽与蛋白结合的亲和力主要取决于两方面:其一是结合界面的焓能贡献(结合能量),其二是由自由态转变为结合态所产生的熵代价。对于在游离状态下能够采取多种构象的肽而言,熵损失尤为显著。为了减少这种熵代价并增强结合能力,研究者广泛采用**交联剂(crosslinkers)**来稳定肽的结合构象(表3)。
最成功的一类交联策略是将肽固定于螺旋构象的“主链订书钉”(helix stapling)技术。该方法通过化学交联将螺旋肽维持在有利于结合的刚性构象中,已被成功应用于多个靶标的结合肽设计,包括MDMX与BCL-2家族蛋白等。目前,研究者已总结出一套相对完善的指南,明确了不同订书钉类型的选择及其在螺旋中的最佳插入位置。
对于含β-折叠(β-sheet)的基序,类似的订书钉体系尚未建立,但已有研究表明二硫键可有效稳定β结构。除了链间交联外,科学家还开发了多种转角模拟结构(turn mimetics)来辅助β-折叠成核,其中最经典的是D-Pro…L-Pro转角基序,已广泛应用于抑制多种靶标的肽设计,如病毒RNA TAR元件、HIV-1 Rev–RRE复合体、CXCR4受体,以及具有广谱抗菌活性的肽。此外,Aib…D-AA或Aib…非手性氨基酸的组合也被证明可作为I’型转角模拟物促进β折叠形成。
并非所有蛋白–蛋白相互作用都依赖二级结构,许多结合是通过所谓的**“热点环”(hot loops)实现的,即界面上承担主要结合作用的柔性环区。研究者已开始利用新型交联剂来稳定这些热点环,以获得更高亲和力的结合肽。Gavenonis等人开发了LoopFinder算法,用于在蛋白–蛋白界面识别这些热点环。随后,他们使用Rosetta软件进行计算性丙氨酸扫描**,评估每个残基的结合自由能变化(ΔG)。当某残基的能量贡献超过1 REU (Rosetta能量单位)时,被定义为热点位点;而同时具有至少三个热点残基(其中两个必须连续),且平均REU≥1的区域被定义为热点环。基于该分析,他们设计出一种能结合stonin2–Eps15相互作用的肽,其亲和力达0.33 ± 0.01 μM。
在另一项研究中,Cerulli等人受天然LC3B结合肽启发,采用非天然氨基酸与多样化订书钉策略,获得了对该靶标具有高亲和力的抑制肽。同样地,London等人提出一种基于Rosetta计算结合能差异的方法,通过比较肽片段与完整蛋白的结合能来识别潜在热点片段。利用该方法获得的肽能够有效抑制多个靶标,包括TLR4、泛素E3连接酶SCF^Fbx4以及gankyrin–ATPase复合物。这一流程已通过Peptiderive服务器实现自动化,输入蛋白–蛋白复合物结构后,系统会自动识别热点片段,并在可能的情况下通过二硫键或N–C端环化进行稳定化。
最常见的天然环状结构之一存在于抗体–抗原相互作用中,这些结合环也可通过交联剂加以稳定以生成结合肽。例如,Kadam等人利用一系列非天然氨基酸,成功稳定了流感血凝素干区结合抗体BnA的环状结构,从而获得了结合亲和力低至17 nM的高效肽分子。
表3 | 交联策略在肽结合物设计中的应用实例。 列举了采用交联(crosslinking)方法稳定肽构象、提升结合亲和力的代表性研究。内容涵盖螺旋订书钉(helix stapling)、二硫键桥接以及环化与转角模拟等多种化学固定手段,展示了它们在增强肽结构稳定性与靶标结合能力方面的应用效果。
2.3 将结构基序嫁接到新支架中
除了交联稳定外,另一种重要策略是**将天然基序嫁接(grafting)到肽或蛋白支架(scaffold)**上(表4)。此方法通过将整个基序或其关键功能基团嵌入一个天然或人工设计的稳定框架中,以提升结构稳定性与耐突变性。
最成功的支架类型之一是富含二硫键的肽类,如结蛋白(knottins)和环肽(cyclotides)。这些刚性极高的支架可容纳环区插入而不破坏整体折叠结构。例如,将整合素结合RGD基序嫁接入结蛋白(Ecballium Elaterium胰蛋白酶抑制剂)并经定向进化后,获得了低纳摩尔级亲和力的整合素结合肽。随后研究表明,工程化结蛋白可用于成像、治疗及药物递送等多种用途。
类似地,将不同受体结合环嫁接入环肽(cyclotides)中,成功获得了多种G蛋白偶联受体(GPCR)的肽结合物,包括缓激肽受体、VEGF-A受体、黑皮质素4受体以及趋化因子受体4等。
除了环区相互作用外,二级结构单元同样可以被嫁接到其他支架上。近年来,一类具有完整折叠结构的小型蛋白(miniproteins)被用于稳定已知能与靶蛋白结合的结构基序。此策略已成功生成对流感血凝素(HA)及肉毒毒素B型等靶标具有极高亲和力与特异性的结合肽。
此外,研究者还将二级结构嵌入更小的肽中。例如,Blosser等人利用β-发夹模拟物稳定延伸的β链,以抑制TCF4/β-catenin相互作用。他们首先确定最小竞争片段(其抑制常数Ki = 57 ± 18 μM),然后在其中加入带有稳定β-发夹的理性设计链段,以增强稳定性、溶解性并产生额外相互作用。最终获得的肽其亲和力提高约10倍(最佳Ki = 4.0 ± 0.5 μM)。
综上所述,交联固定与支架嫁接为肽的结构稳定化提供了两条互补路径,使研究者得以在提高亲和力的同时保持肽分子的构象完整性,为高性能结合肽的设计奠定了基础。
表4 | 不同结构基序嫁接至现有支架的实例。 汇总了将多种天然或人工结构基序(motifs)嵌入稳定肽或蛋白支架(scaffolds)的代表性研究,展示了通过环肽、结蛋白、迷你蛋白等刚性框架嫁接螺旋、β折叠或环状结构以提升肽分子的稳定性、亲和力与特异性的典型应用。

3 从头设计功能性肽
顾名思义,从头设计(de novo design)是指完全从零开始构建肽分子的过程。不同文献对该术语的定义略有差异,该综述中,“从头设计”特指那些并非基于天然蛋白界面中已知基序进行稳定化的肽设计方法。当前从头生成功能性肽的策略可分为三类(图4):
(a) 先生成序列,再进行构象建模/采样;
(b) 先生成结构,再进行序列优化;
(c) 同时对序列与结构进行采样。
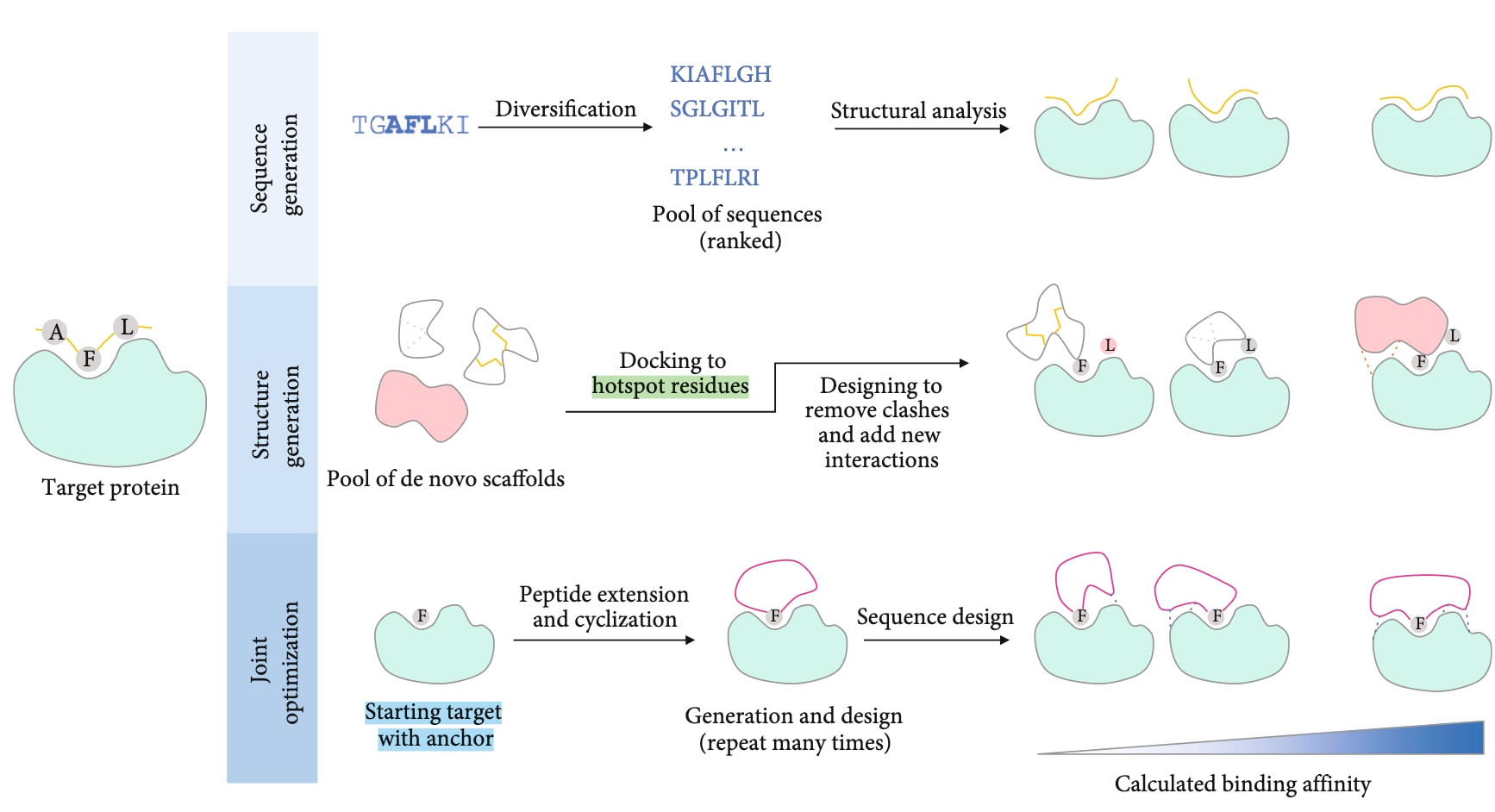
图4 | 从头设计生成肽结合物的方法概览。 从头设计肽结合物可大体分为三类策略:(a) 从头生成序列:先产生多样化的序列库,再基于序列评分及下游结构分析的结合能计算筛选出最优结合肽;(b) 从头生成结构:首先构建具有特定构象的结构支架(scaffold),用于在界面上呈现并稳定一组关键热点残基(hotspot residues);(c) 序列与结构联合优化:在同一步骤中同时生成序列与结构。该方法通常以界面处的**锚点(anchor)**功能基团为起点,在靶蛋白口袋环境中生成肽结构,优化序列并根据序列调整构象,以获得最佳结合体。多轮循环后可形成一组高潜力结合肽候选。
3.1 从头生成肽序列
基于序列的设计方法(图5)通常从已知结合肽或一组结合序列库出发,通过引入多样性来筛选最优结合肽。由于肽序列空间极其庞大,这类方法往往在生成时施加一定约束条件,或引入优化算法以缩小搜索范围。约束类型与优化策略通常取决于研究者掌握的信息及靶标特征。生成的候选序列随后会被打分与排序,以挑选最具潜力的结合肽。这种排序可通过能量计算、对接模型、或结合结构预测实现。
Smadbeck等人以设计选择性抑制EZH2的肽为例,构建了一个多阶段的设计框架。首先,从天然EZH2结合肽出发生成一系列候选序列,并通过能量最小化筛选出稳定构象;随后评估这些序列能否重现模板肽的空间折叠特征;再使用结合能计算方法(RosettaDock)对前45个候选进行筛选,并最终通过实验验证。多数预测肽显示出低微摩尔(μM)级的抑制活性,其中设计肽SQ037的IC₅₀为13.57 μM,虽低于现有纳摩尔级小分子抑制剂,但验证了方法的有效性。
Unal等人采用了类似的多步式从头设计框架。首先利用高斯网络模型(Gaussian Network Model)确定靶蛋白的结合位点,然后使用Autodock在该位点上对接一条肽(若缺乏天然配体,则使用poly-Ala肽)。接着计算所有可能氨基酸对在结合界面的结合能,并通过基于马尔可夫模型的转移概率预测最优序列。该方法在多种靶标上得到验证,包括HIV-1蛋白酶与scytalidocarboxyl肽酶B,实验结果表明其设计的肽结合能力显著增强。例如,算法设计的三肽Trp–Tyr–Val对HIV-1蛋白酶的结合能为**−9.59 kcal/mol**,明显优于天然抑制肽Glu–Asp–Leu (−7.66 kcal/mol)。
Li等人则提出了一种用于抑制VEGFR-3的肽设计方法。他们首先利用FTMap算法确定VEGFR-3蛋白表面的结合口袋,并基于Mekler–Idlis“正反义氨基酸配对理论”构建了576条肽序列库(图6)。随后通过分子对接与构象分析对这些肽进行排序,并合成高评分序列进行实验验证。结果表明,设计肽CP-7 (序列CVKTFDP)在细胞实验中对A549细胞的结合率高达94.6%,远高于阳性对照肽LARGR的57.8%。
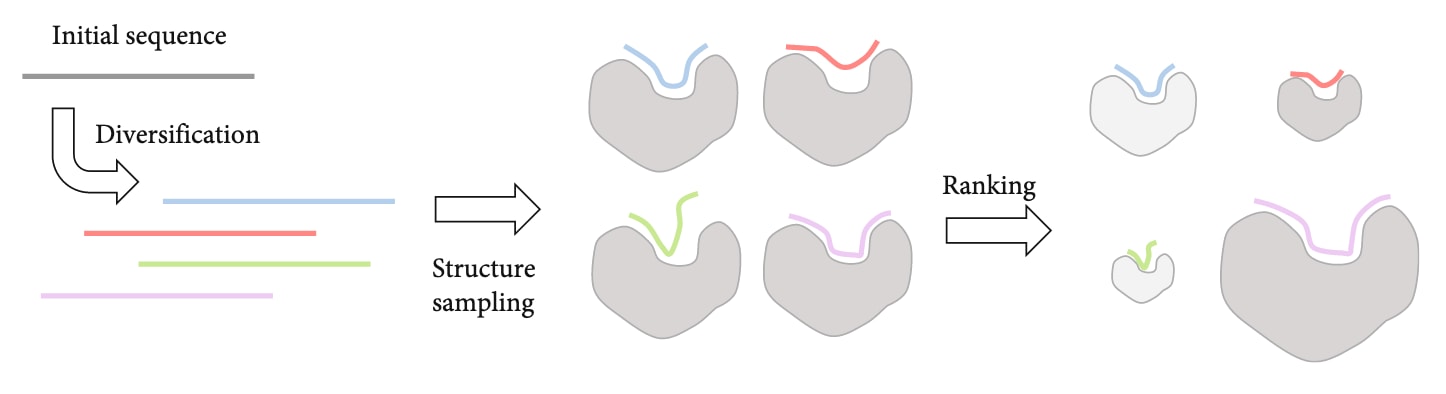
图5 | 基于序列采样的从头设计方法总体示意图。 该设计流程首先经过序列多样化步骤,生成一组候选肽序列;随后进行结构建模与打分,根据模型质量对序列进行排序。右侧示意中,圆形大小与序列排名成正比(越大代表模型评分越高)。最终,得分最高的序列将被用于实验验证,或作为下一轮设计与优化的起始序列输入。
3.2 从头生成肽结构
在此策略中,研究者首先设计具有预定义三维结构的肽,再将其作为支架(scaffold)用于靶标结合(表5)。这些支架通常包含若干关键热点残基(hotspot residues),以确保结合能力。设计的关键在于使热点残基呈现出有利于结合的空间取向。与第二章中通过交联或嫁接整个二级结构的方式相比,该方法更具结构主导性。
在某些相互作用中,仅需正确呈现关键侧链即可实现结合,尤其是在界面由α螺旋或β折叠主导时。基于此思想,研究者开发了一系列能够形成稳定α螺旋或β折叠构象的拟肽样折叠体(foldamers),其主链趋向可预测,从而能以特定方式展示侧链。由于蛋白界面上螺旋基序极为常见(图7),α螺旋模拟物的设计成为从头构建结合肽的主要方向之一。
例如,肽–寡脲杂合物(peptide–oligourea hybrids)被用作α螺旋模拟物,成功抑制了MDM2与维生素D受体等靶标;三联苯(Terphenyl)支架也被用于模拟螺旋并阻断Bcl-xL/Bak相互作用。此外,β-发夹(β-hairpin)亦能模拟螺旋暴露残基的排列。Fasan等人利用小型β-发夹稳定p53–HDM2界面的两个热点残基,通过多轮突变优化后获得100 nM级亲和力的结合肽。
另一类常用支架是从头设计的α螺旋卷曲(coiled-coil)。Fletcher等人构建了平行与异源二聚α螺旋结构,用于干扰MCL-1/NOXA-B界面。研究者首先通过Robetta计算性丙氨酸扫描确定关键残基,并将其分为“短基序”和“扩展基序”,再嫁接到从头设计的CC-Di肽上,生成多种混合构型。实验验证显示这些肽能有效结合MCL-1并稳定卷曲结构。
热点残基还可计算生成。例如,Cao等人以小型蛋白(miniproteins)为支架,整合计算设计的热点残基,获得了对SARS-CoV-2刺突蛋白的抑制剂,其IC₅₀低至5 pM。另一种无热点策略则从形状互补性出发:Schneider等人针对TCF/β-catenin相互作用,从高分辨率的肽样或肽–拟肽杂合支架中筛选出最优结构,通过计算突变与实验筛选,获得了IC₅₀ = 5.44 ± 0.82 μM的结合肽,并在细胞中验证了抑制效果。
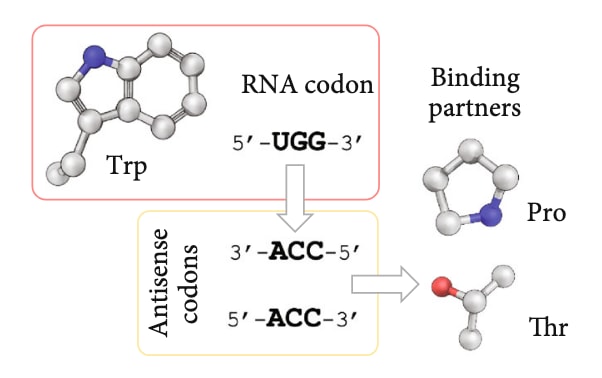
图6 | 正反义氨基酸配对理论示意图。 该理论的核心假设是:在为肽设计选择与靶标表面结合的氨基酸时,与界面上氨基酸呈反义(antisense)关系的残基,其结合亲和力通常高于随机选择的氨基酸。两种氨基酸是否构成正义(sense)与反义(antisense)配对,是根据其tRNA密码子之间的互补关系来确定的,如图所示。
表5 | 用于结合蛋白靶标的从头设计支架实例。 该表汇集了多种从头构建的肽或蛋白支架(de novo designed scaffolds)在靶蛋白结合中的应用实例,涵盖螺旋、β-折叠及混合折叠结构等类型。这些支架通常通过热点残基的空间定向呈现或骨架刚性优化实现高亲和结合,展示了结构驱动型设计在功能性肽开发中的潜力。
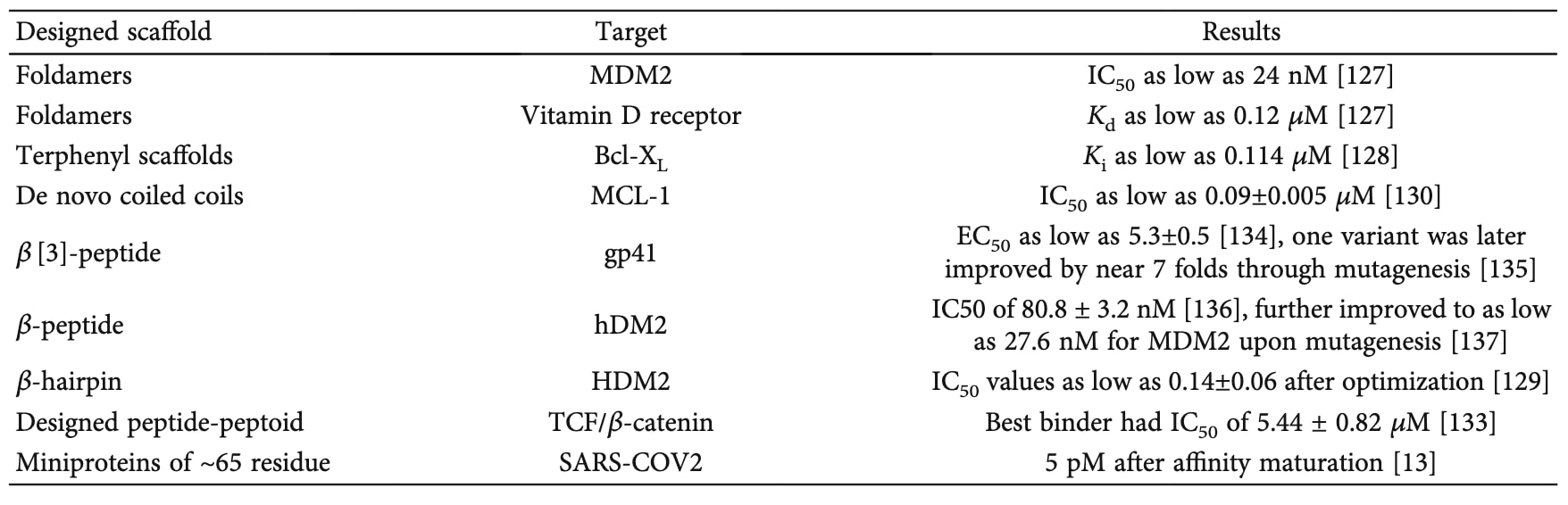
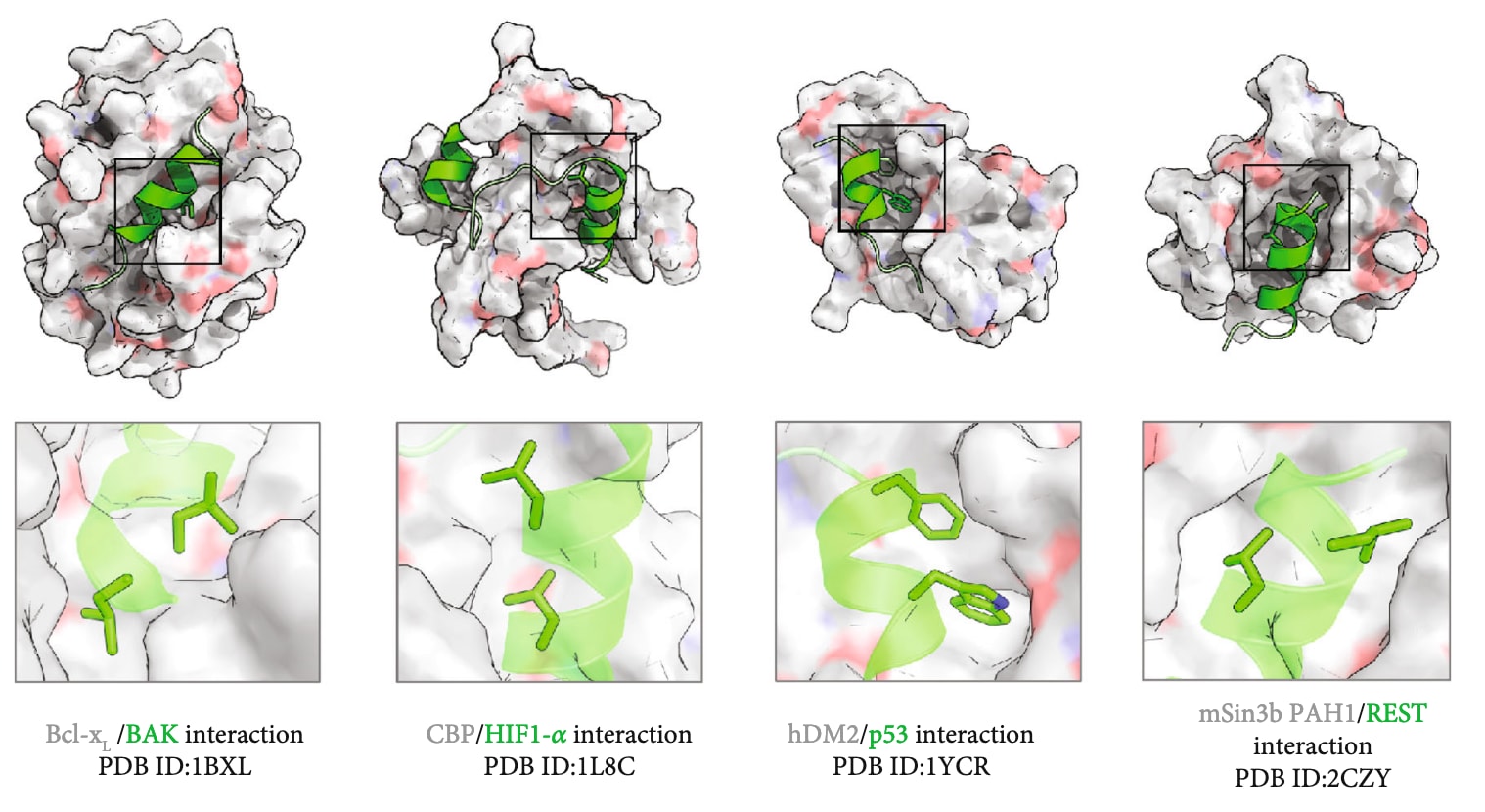
图7 | 螺旋基序在蛋白–蛋白界面中普遍存在。 图中展示了多种由α螺旋介导的蛋白–蛋白相互作用实例。棒状结构表示在结合中起关键作用的热点残基(hotspot residues);下方各框为上方总体结构中热点区域的放大视图。底部面板中螺旋以半透明示意方式呈现,以便清晰显示界面中热点残基的空间分布。
3.3 序列与结构的联合优化
序列与结构的同步优化(simultaneous optimization)是最直观也是最理想的设计思路,但在计算上往往极其昂贵。分子动力学(MD)模拟虽然能捕捉结构变化,却需大量计算资源,而在模拟中同时引入突变几乎不可行。某些从头设计方法允许在固定主链的基础上进行微小骨架移动以稳定结构,但由于单一刚性主链的序列优化本身已属高复杂度问题,因此难以引入MD所表现的那种大尺度构象变化。
然而,对于非常短的肽,两种过程可以结合使用,从而设计出具有预定义结构或功能的肽分子。线性肽因其柔性过高而难以准确预测,但受限肽(constrained peptides)——如环肽(cyclic peptides)——由于自由度较小,可通过计算方法设计出预设构象。
2018年,Slough等人采用分子动力学(MD)在水环境中设计出具有刚性结构的环状五肽(pentamers)。他们利用BE-META MD方法对一系列仅在两个残基不同(除Pro外)的环五肽进行模拟,并基于模拟结果建立了用于预测环肽结构的打分函数。该评分依据不同二肽对在模拟中倾向形成的构象偏好计算。其中一个肽在模拟中形成了稳定结构,并被NMR实验证实;但多数序列未形成稳定构象。研究者发现,一些转角组合在打分中表现较差,因此认为仅通过常规氨基酸替换难以获得理想结构,并建议引入**非天然氨基酸(ncAAs)**以稳定特定构象,并提出该方法可扩展至更大环肽或其他类型环键体系。
除了MD方法外,Rosetta软件也被广泛用于环状肽的结构设计。该方法利用一种名为广义运动链闭合算法(GenKIC, generalized kinematic loop closure)的计算策略(图8),该算法灵感来源于机器人学中的运动约束计算,用于预测在约束条件下可实现的构象空间。通过GenKIC,研究者从多种poly-Gly骨架出发采样大量环肽主链构象,并在聚类后为其分配侧链:将D-型氨基酸限定于φ>0的位置,L-型氨基酸限定于φ<0的位置;同时排除甘氨酸以避免过度柔性。这样可确保侧链取向能够稳定主链折叠。NMR研究表明超过70%的设计肽确实采用了预测构象,显示了该方法的高成功率。利用该策略生成的7–10残基环肽库中,计算识别出超过200种稳定结构。与MD方法相比,Rosetta的优势在于速度更快、覆盖范围更广,可在11个氨基酸长度范围内几乎穷举所有D/L组合。
此外,该算法还可与其他设计模块结合,用于在靶蛋白表面同时采样序列与构象,生成靶向结合肽。具体流程如下:以已知结合分子或锚点(anchor)为起始点,在靶标口袋中扩展并环化骨架;随后利用GenKIC采样肽构象,并为每种构象设计序列以实现最大化相互作用;经侧链与构象精修后,根据界面形状互补性、结合自由能ΔG等指标筛选模型,最终挑选最佳候选用于实验验证。
该方法已成功应用于多种治疗性靶标:
- PD-1受体抑制肽(Kd = 30 μM 与 102 μM),可与天然配体竞争;
- 抗生素耐药因子NDM1抑制肽,其中6/7候选的IC₅₀低于D-captopril对照,最佳肽IC₅₀ = 1.2 ± 0.1 μM,比对照低约50倍;
- HDAC-2与HDAC-6去乙酰化酶抑制肽中,17/39显示IC₅₀≤100 nM,最佳结合物对HDAC-6与HDAC-2的IC₅₀分别为约4 nM与9 nM,比起始锚点提升约三数量级。
这些研究总体遵循相同的计算流程,但在细节上各有改进。例如:
- Mulligan等人引入了新的能量项,不仅依据整体评分,还考虑界面质量,并指出更刚性的肽具有更高结合性能,因为柔性肽具有更大焓变,对ΔG不利;
- Hosseinzadeh等人则发现,即使无稳定结构,无序肽也能获得高亲和结合。他们在靶口袋附近加入局部穷举搜索以识别最优侧链接触,大幅提高了成功率。
总体而言,序列与结构联合优化策略通过在一个统一框架中协调主链构象采样与序列能量最优化,已成为环肽与受限肽设计的核心手段,在实现纳摩尔级甚至皮摩尔级结合亲和力方面展现出极高潜力。
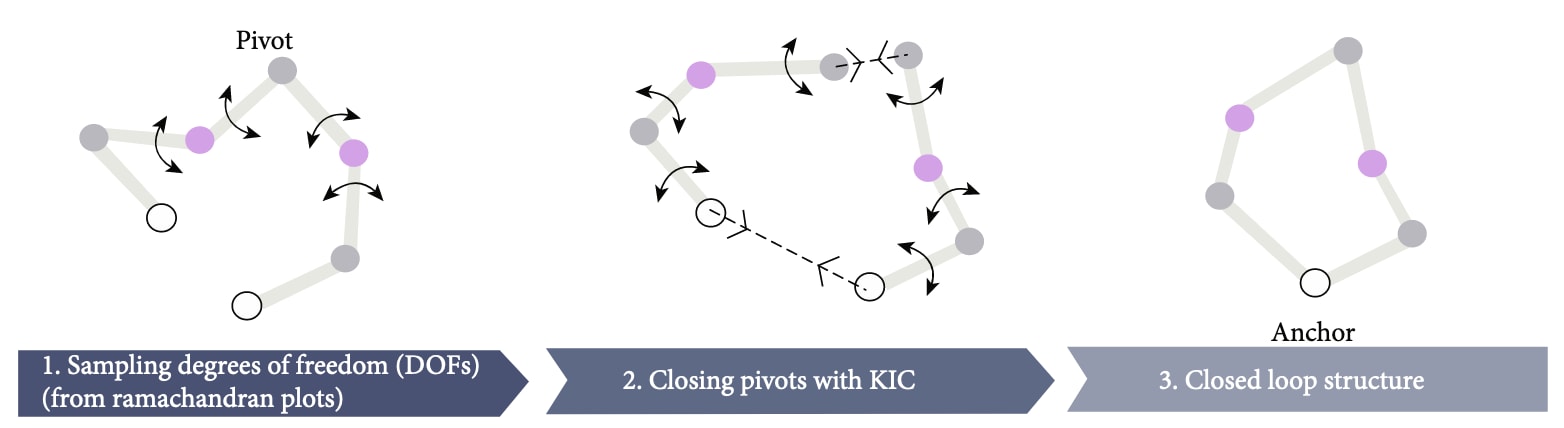
图8 | 利用广义运动链闭合算法(GenKIC)采样环状肽的示意图。 该方法的第一步是随机采样除三个“枢轴点”(pivot residues)外所有残基的扭转自由度;第二步则计算这三个枢轴残基的二面角,以确保形成闭合环结构。整个过程中,**锚点(anchor)**的位置保持不变,从而在约束条件下生成符合空间闭合要求的环肽构象。
4 用机器学习设计功能性肽
尽管深度学习的最新进展已经彻底革新了蛋白质结构预测领域,但在预测肽的结构或功能,以及设计具有新功能的肽方面仍面临巨大挑战。该节首先回顾机器学习方法在肽结构与功能预测中的成功应用(见4.1节),随后介绍其在新肽设计中的最新进展(见4.2节)。
用于肽设计与预测的学习方法高度依赖于肽的表示方式及训练集中可用数据的类型与规模(见图9)。因此,该节重点涵盖多种肽表征策略与学习算法,以全面展现该领域的研究格局。
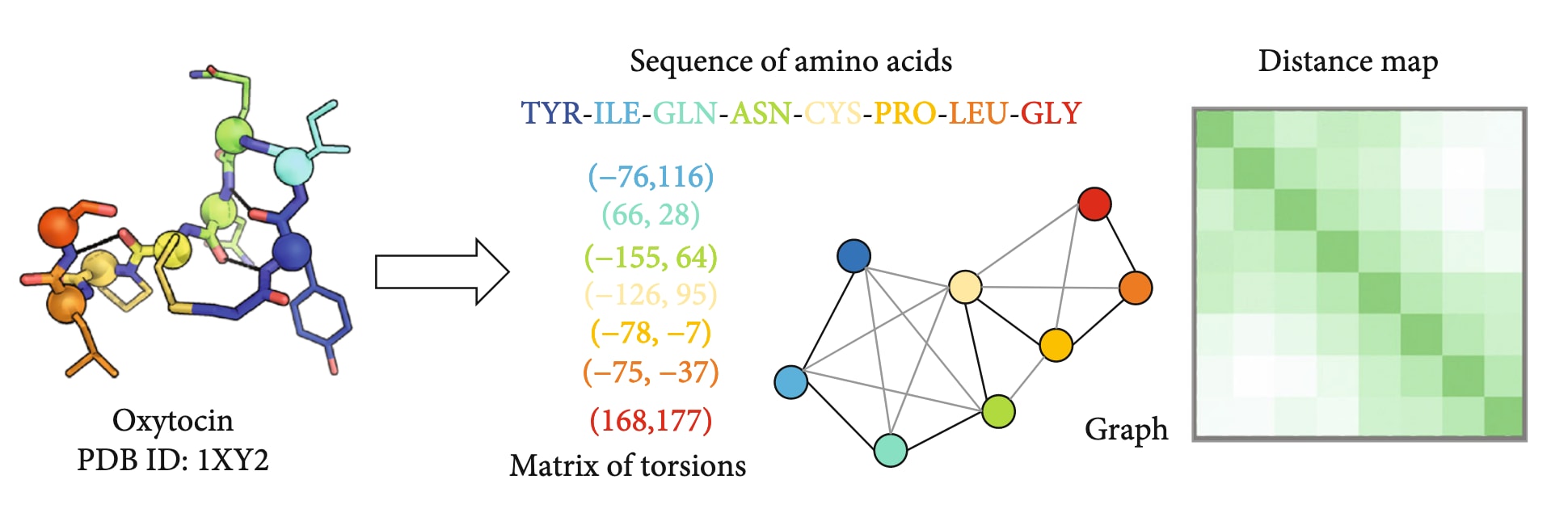
图9 | 肽分子可通过多种方式进行表示。 不同的表示方法反映了肽分子的不同特征。例如,序列级输入仅包含氨基酸顺序信息,而不显式体现其结构特征。针对每一种输入表示,研究者可选择不同类型的机器学习算法进行建模与预测,以捕获序列、结构或理化性质层面的信息。
4.1 预测方法
4.1.1 结构预测
近年来,计算算法的发展使得环状肽的结构设计取得显著进步,可实现稳定单一构象的肽分子(详见第3节)。然而,许多天然肽在溶液中能呈现多种构象,预测这些构象对于设计具有特定功能的肽片段至关重要。然而,准确预测环状肽在溶液中可采样的多种构象仍具挑战。
一个重要进展是StrEAM算法,它将分子动力学(MD)模拟与深度学习相结合以预测肽的构象。Miao等人通过显式溶剂模型的偏置交换势垒增强动态法(bias-exchange metadynamics),捕捉了环肽在溶液中的转动行为。相比不考虑溶剂的简化模型,显式溶剂模型能更准确地描述主链C=O与N–H键与水分子形成的氢键笼结构。
在该算法中,研究者将五肽的结构以每个残基的
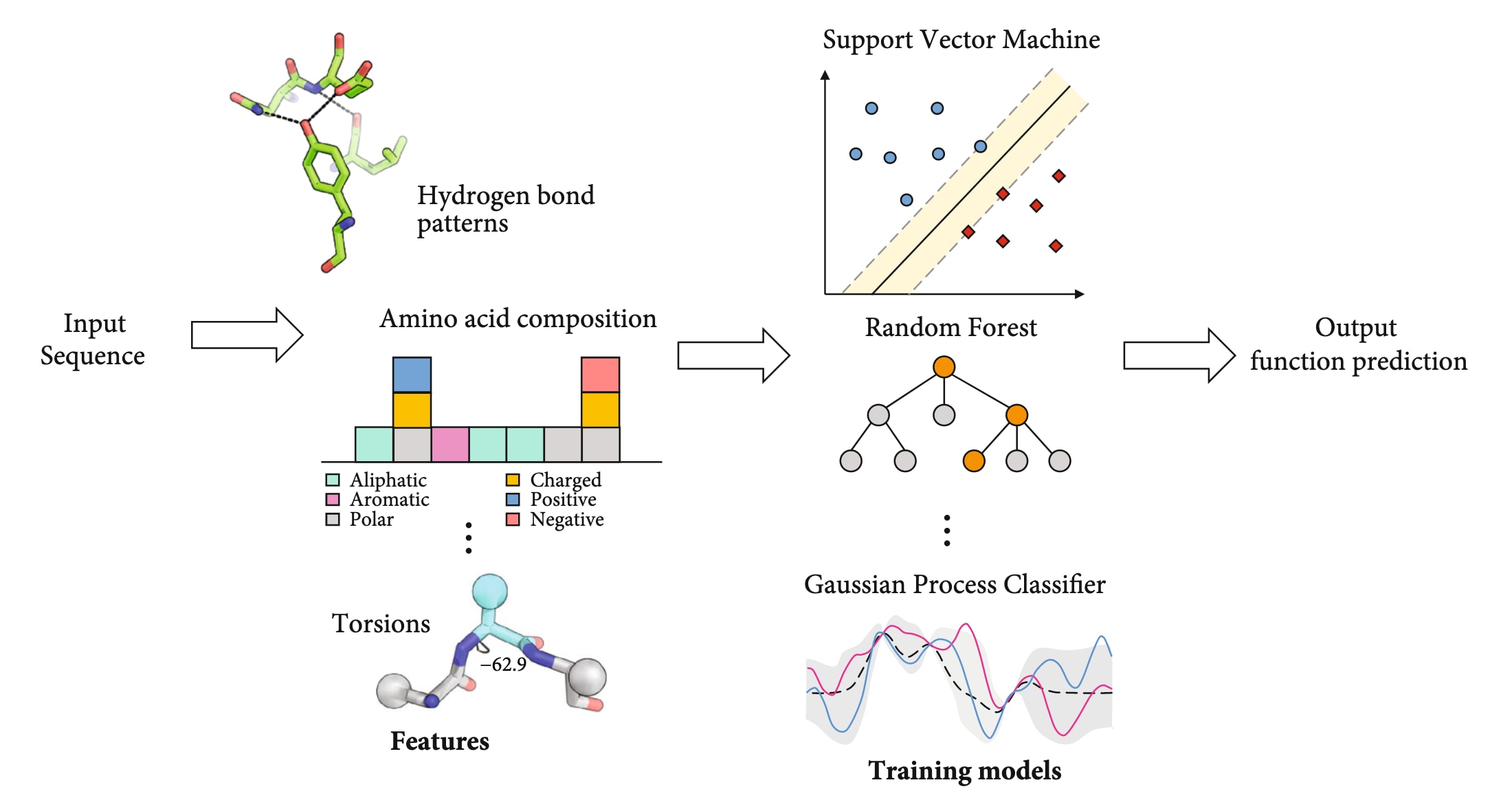
图10 | 肽序列细胞穿透能力预测的一般流程示意图。 预测算法首先输入待测肽序列,随后从中提取多种特征,包括氨基酸组成、可旋转键数量、氢键数量、扭转角信息及其他理化性质等,用于训练预测模型(如支持向量机SVM、随机森林、高斯过程分类器等)。训练完成后,模型即可对输入序列的细胞穿透能力进行预测与评估。
4.1.2 功能预测
除结构预测外,概率模型、分子建模与深度学习也被广泛用于筛选具有生物活性的肽并预测其功能。以下介绍几个代表性应用。
(1) 细胞穿透肽(Cell Penetrating Peptides, CPPs)的预测
细胞穿透能力是肽分子的一项关键功能。为此,研究者已开发出多种CPP预测器(图10),并可扩展用于其他肽功能预测(详见表6)。
Gautam等人开发了网络平台CellPPD,利用支持向量机(SVM)预测并设计CPP。其模型基于来自CPPsite数据库的708条CPP序列,并结合氨基酸组成、二肽组成与理化性质等特征。
除SVM预测器外,CellPPD还引入了MEME/MSAT基序识别算法的混合模型,预测准确率高达97.4%,优于单独使用SVM或MEME/MSAT的结果。研究还发现,Arg、Lys与Trp残基在CPPs中显著富集,并在序列不同位置分布规律性不同。此外,CellPPD可对用户输入序列进行点突变预测,建议具有更强穿膜能力的变体。
Wei等人进一步提出改进版预测器SkipCPP-Pred,基于k-skip-n-gram语言模型提取序列特征并训练随机森林(random forest)分类器。该方法借鉴自然语言处理中的上下文预测机制,可捕获残基间关联,预测准确率为90.6%,优于CellPPD-DC (87%)与CellPPD-BP (83.7%)。
Qiang等人开发的CPPred-FL采用随机森林算法,并引入特征学习与优化步骤,将高维特征空间映射至低维空间。使用前19个最优特征时,预测准确率达到92.1%。
Fu等人在2019年提出另一种基于SVM的CPP预测模型,利用氨基酸频率与理化组分作为特征,训练后准确率达92.3%,优于CellPPD与SkipCPP-Pred。
de Oliveira等人则构建了BChemRF-CPPred框架,可区分CPP与非CPP。该系统整合神经网络(MLP)、SVM与高斯过程分类器(GPC),并采用投票集成机制输出最终预测结果。
与多数仅基于序列特征的模型不同,BChemRF-CPPred引入了结构描述符,包括分子量、可旋转键数、拓扑极性表面积、净电荷与带负电残基数等,与膜通透性直接相关。结构特征的引入使准确率由86.5%提升至90.66%,在独立数据集上(60个CPP与75个非CPP)的准确率达89.62%,显著优于CPPred-RF(68.88%)与SkipCPP-Pred(62.58%)。
表6 | 部分用于预测细胞穿透肽(CPP)功能的方法概览。 该表汇总了多种CPP预测模型的主要特征与性能指标,包括其所采用的算法类型(如SVM、随机森林、深度学习等)、特征输入(序列或结构特征)及预测准确率。需注意,文中所列准确率来自不同数据集,不可直接进行横向比较。

(2) 抗癌肽(Anti-Cancer Peptides, ACPs)的预测
肽因其高特异性、低毒性与良好膜渗透性,被广泛探索用于癌症治疗。然而,抗癌肽的实验数据有限,严重制约模型构建。
为此,Chen等人提出深度学习模型xDeep-AcPEP,可预测肽对六种肿瘤细胞(乳腺、结肠、宫颈、肺、皮肤、前列腺)的抗癌活性。该方法采用多任务学习(MTL)策略,为不同组织独立训练多个卷积神经网络(CNN)。
模型使用CancerPPD数据库中经实验验证的抗癌肽作为训练集,并结合AAINDEX、BLOSUM62、ZScale(反映氨基酸疏水性、体积、电性等理化属性)以及独热编码进行序列表示。网络结构包含卷积层+ReLU激活、平均池化、批归一化与最大池化层。
实验表明,MTL模型较单任务模型(STL)具有更强泛化能力,并能准确预测高活性肽的抗癌性,但对低活性或无活性肽的预测仍有限。
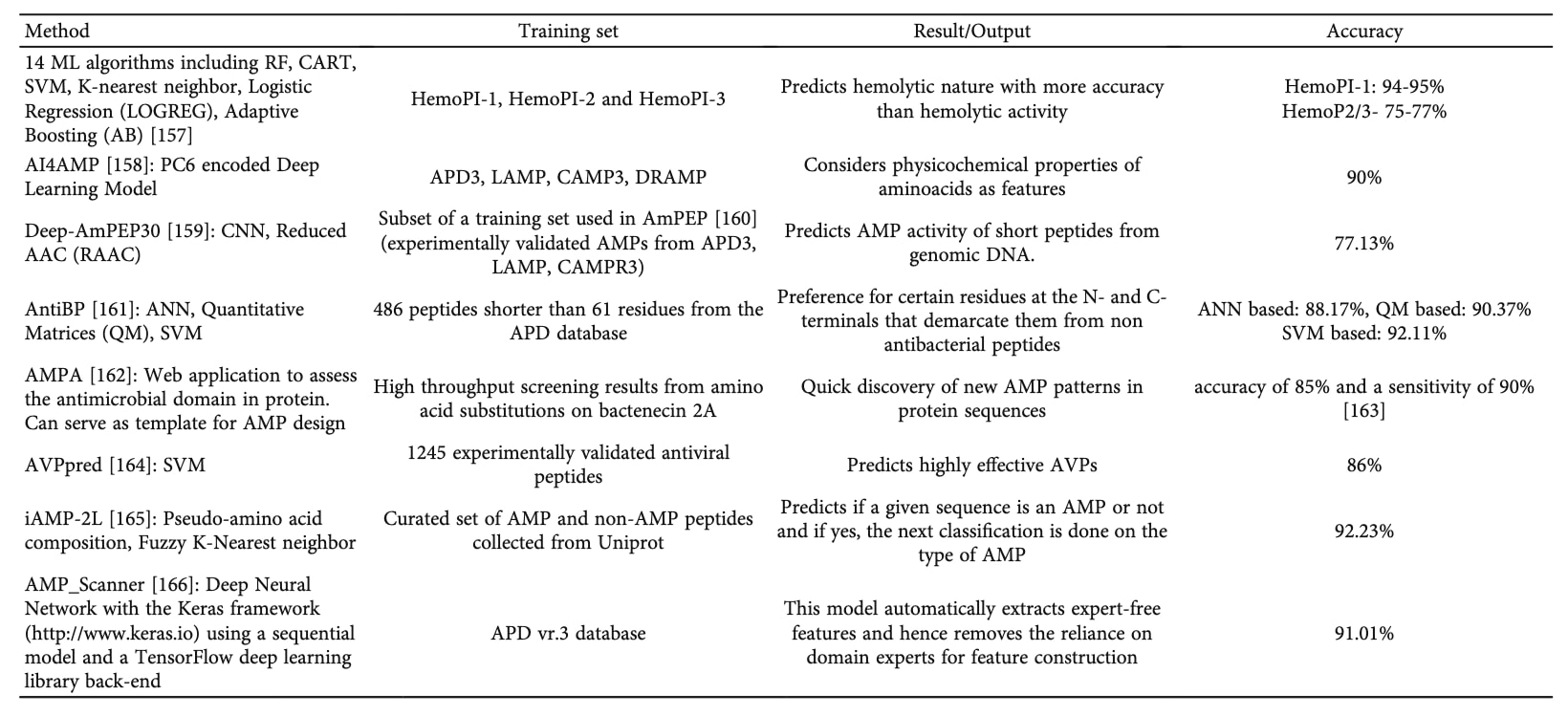
(3) 抗菌肽(Antimicrobial Peptides, AMPs)的预测
抗菌肽是替代抗生素以应对耐药感染的潜在方案。近年来,从传统机器学习到深度学习的多种AMP预测模型不断涌现(见表7)。
AMP研究的关键问题之一是其溶血性(hemolysis),即对红细胞膜的破坏。为降低毒性,Plisson等人开发了基于14种算法(包括CART、随机森林与SVM)的ML模型,并在HemoPI-1、HemoPI-2、HemoPI-3数据集上训练。模型以56个理化特征(芳香性、疏水比、净电荷、体积等)为输入,并在APD数据库的3081条天然AMP上测试,能准确识别溶血性肽。
Lin等人构建了网络平台AI4AMP,提出PC6蛋白编码方法,融合疏水性、侧链体积与等电点等特征,预测精度达90%。
Yan等人开发了Deep-AmPEP30,基于深度卷积网络预测基因组来源短肽的抗菌活性。模型在真菌Candida glabrata基因组中预测出一条20个氨基酸的活性肽,并通过实验验证了其抗菌作用。
表7 | 抗菌肽(AMP)活性预测方法概览。 该表总结了多种用于预测AMP活性的计算方法,包括传统机器学习模型与深度学习模型,并列出其主要算法类型、输入特征(如氨基酸组成、理化性质或序列编码方式)及预测性能指标。需强调,表中列出的准确率基于不同基准数据集,因此结果之间不可直接比较。
(4) 肽–蛋白相互作用(Peptide–Protein Interaction, PPI)的预测
许多肽的功能来源于其与蛋白伙伴的结合,因此预测肽–蛋白相互作用具有重要意义。
Lei等人提出了深度学习框架CAMP,可预测肽与蛋白是否形成复合物。CAMP基于PDB中的肽–蛋白复合物与DrugBank中的配对信息,使用PLIP工具提取非共价相互作用数据,并结合PepBDB数据库标注参与结合的残基。
模型提取肽与蛋白的结构与理化特征,并输入卷积神经网络(CNN)与自注意力模块(self-attention),同时预测是否结合以及哪些残基参与作用。其平均AUC为0.803,MCC均值为0.489。模型成功预测了已知复合体,如GLP-1受体与Semaglutide肽配体,展示了在肽–蛋白相互作用预测中的潜力。
综上,机器学习正在从功能预测逐步走向功能生成,通过结合结构建模、理化特征与神经网络表达,为功能性肽的可解释性设计与高通量筛选提供了强大支持。
4.2 生成式方法
在功能性肽设计中,基于学习的方法常受到训练数据不足的限制。为克服这一问题,研究者提出了多种创新思路,将**(半)高通量实验数据生成与机器学习相结合,从而加速肽的设计流程。事实表明,即便只从具有代表性的小型数据子集中学习,也能显著提升设计方法的成功率。
该节首先介绍结合实验与学习的“设计–测试–学习(Design-Test-Learn)”类方法(4.2.1节),随后讨论基于数据库的全计算生成模型(4.2.2节)。
不同模型的输入形式与学习策略依赖于目标功能与可用数据类型,因此该节的重点在于涵盖多种数据表示与建模方式。相关研究实例汇总见表8**。
4.2.1 设计–测试–学习方法
Jenson等人结合实验与计算策略,生成了对三种靶标(Bcl-xL、Mcl-1与Bfl-1)具有高亲和力与高选择性的功能性肽。研究团队首先采用一种高通量筛选技术——amped SORTCERY,测定约1万个肽(来自已设计的组合文库)对上述蛋白的结合亲和力。随后,他们使用支持向量回归(SVR)对实验数据进行拟合,并将模型应用于约2760万条肽序列以及更大的候选集(约
Tallorin等人开发了一种计算–实验混合方法POOL(Peptide Optimization with Optimal Learning),用于快速优化具有特定生化功能的肽。POOL基于朴素贝叶斯算法,根据实验数据学习每条序列成为“命中(hit)”的概率。初始训练集包含针对两种目标酶(Sfp与AcpS)的活性肽与非活性肽。每轮实验后,新数据都会并入训练集,并重新训练模型,使其预测出“在给定实验轮次下至少出现一个新命中序列的概率最大”的候选肽。该模型在预测新命中肽时假设此前推荐的肽为非命中序列,从而避免局部最优。
经过四轮迭代学习与实验筛选,研究者获得了可选择性标记AcpS或Sfp的短肽。POOL模型通过平衡“探索(exploration)”与“利用(exploitation)”显著提高了有效肽的采样效率。
Schissel等人将高通量实验与深度学习相结合,从零设计出人工合成(abiotic)核靶向迷你蛋白(miniproteins)。他们合成并测试了600个PMO(磷酰胺吗啉寡聚物)–肽偶联体,并利用实验活性数据训练卷积神经网络(CNN)预测器。生成器部分采用嵌套LSTM结构,并结合遗传算法进行序列优化,最终获得能高效穿透细胞膜的新型蛋白。模型预测的多种序列与实验活性高度吻合,表明其生成机制具有较强的真实性与泛化能力。
4.2.2 全生成式模型
从头设计抗癌肽(ACP)是生成式AI在肽药物设计中的典型应用之一。
Grisoni等人提出了一种反向传播人工神经网络(CPANN)模型,用于生成具有抗癌活性的肽。研究者从CancerPPD数据库手动整理出针对乳腺癌与肺癌的肽序列,用作训练数据,并通过pepCATS描述符提取其药效团特征。CPANN模型由Kohonen自组织映射层与Grossberg输出层组成,可自动聚类未知功能肽。该模型被应用于由modLAMP工具包(一种用于肽设计与可视化的Python程序)生成的1000条α螺旋肽,以预测其抗癌潜力。
在合成并测试的18条肽中,6条显示抗癌活性,其中5条能同时作用于两种癌细胞系(MCF7与A549),表明模型具有一定的多靶点预测能力。
另一项研究中,研究者提出一种基于双向LSTM(Bi-LSTM)的序列生成模型,用于构建与代表性肽结构相似的序列。每个氨基酸被视为一个“词元(token)”并向量化输入网络。模型由两层Bi-LSTM组成(每层含1024个隐藏单元),在Pfam数据库(2100万序列)与SCOP数据库(2万序列)上训练。该模型能重建蛋白序列并最小化交叉熵损失,从而预测蛋白的二级结构、结构相似性与接触图(contact map)。在SCOP结构分类中准确率达91.2%。
研究者进一步将该模型用于几丁质结合肽的设计。以poly-Ala序列为起点,通过突变生成序列库,并根据模型预测的结构相似度筛选,循环迭代至相似性得分>3.5。筛选出的候选肽经分子动力学模拟与生化实验验证,表现出与天然肽AC2-WT相似的抗真菌活性。
Capechhi等人利用机器学习设计非溶血性抗菌肽(AMPs)。他们的模型结合了预测型与生成型循环神经网络(RNN),以DBAASP数据库中的带注释序列(含活性与溶血信息)为训练集,并通过迁移学习(transfer learning)在特定菌株(P. aeruginosa, A. baumannii, S. aureus)上进行微调。随后,模型生成新肽序列并依次通过AMP活性分类器与溶血性分类器筛选。最终合成并测试了28条短肽(≤15个残基),其中64%表现出MIC ≤16 μg/mL的抗菌活性,且约半数肽同时具备活性与非溶血特征。
综上所述,生成式学习方法正从“预测”迈向“创造”。通过结合实验反馈、高通量筛选、深度神经网络与概率优化,研究者已能在庞大的序列空间中自动探索并生成具备目标功能的肽分子,这标志着AI驱动的肽药物从设计到验证的闭环体系正在逐步成形。
表8 | 基于学习的功能性肽设计方法概览。 该表总结了多种机器学习与深度学习方法在功能性肽设计中的应用实例,涵盖从预测型模型(如SVM、随机森林、CNN)到生成式模型(如RNN、变分自编码器、强化学习)等多种框架。表中列出了各方法的输入特征、训练数据来源、设计目标与代表性成果,以展示当前学习驱动肽设计的主要思路与发展趋势。
表8 | 基于学习的功能性肽设计方法概览。 该表总结了多种机器学习与深度学习方法在功能性肽设计中的应用实例,涵盖从预测型模型(如SVM、随机森林、CNN)到生成式模型(如RNN、变分自编码器、强化学习)等多种框架。表中列出了各方法的输入特征、训练数据来源、设计目标与代表性成果,以展示当前学习驱动肽设计的主要思路与发展趋势。
5 未来展望
功能性肽与蛋白片段的设计为解析与干预蛋白–蛋白相互作用以及开发新型治疗分子提供了令人振奋的机会。从天然结合基序的稳定化,到界面新序列的从头设计,再到深度学习驱动的生成策略,研究者已探索出多层次的方法体系。
以天然基序为起点的结构引导型设计已经成功产出大量高亲和力肽结合物,其中部分成果已促成基于肽结合物的生物技术公司成立;同时,该方向也推动了蛋白设计领域的多项技术进步,如非天然氨基酸参数化、环肽构象采样与预测、以及从稀疏数据中学习的机器学习模型。然而,基于天然结构的策略天然受到“已知结构依赖性”的限制,仅适用于已有结构模板的体系。此外,所得肽的亲和力高度依赖于起始基序本身的结合能力,往往还需经历多轮突变与优化才能获得可与天然结合物竞争的肽分子。
近年来,研究重点正逐渐转向自动化计算与机器学习驱动的肽设计与功能预测。尽管基于Rosetta的设计方法与分子动力学(MD)模拟在多项研究中取得成功,但这些方法仍受限于能量函数(score function)的准确性。计算所得界面能量指标往往无法完全解释实验结果,结合模式的预测依旧极具挑战。未来,改进评分函数与提升采样效率将是增强计算设计能力的关键。
基于学习的肽预测与设计方法仍处于早期阶段,其最大瓶颈在于实验数据的匮乏。要充分发挥这些方法的潜力,需要同时推进学习算法的技术革新与训练数据的扩充。结合高通量文库筛选与机器学习的策略将尤为重要,因为这类方法能产生大规模、带标签的功能数据集。例如,将计算设计的肽与噬菌体展示(phage display)或mRNA展示等文库筛选技术相结合,可构建针对性更强的筛选文库,并获得更具信息价值的实验结果。
与此同时,蛋白结构预测的突破为肽结构与复合体预测带来了全新契机。若能实现蛋白–肽复合物的高精度建模并结合结构层面的亲和力预测,将对肽结合物的理性设计产生变革性影响,尤其在选择性结合肽的设计领域——这一目前仍缺乏普适策略的方向。
值得强调的是,大多数肽结合物的最终目标是应用于治疗开发。结合仅是药物研发流程的第一步,许多高亲和力候选在后续开发中因药代动力学性质不理想、体内半衰期短或毒性过高而失败。此外,目前的结合实验往往忽略了细胞穿透性测试,使得某些高亲和力肽无法进入细胞结合目标蛋白。因此,预测肽的膜通透性与药代动力学特征将成为未来研究的重要方向。
由于缺乏这些性质的大规模数据,研究者需要开发能够从噪声大、失败率高的数据中提取有效信息的新特征与新方法。可以预见,未来的功能肽设计将融合结构预测、生成式学习、高通量验证与药理建模,构建一个从序列到功能再到药性优化的闭环式智能设计体系,推动肽类药物的精准与可编程化设计迈向新阶段。