Acc. Chem. Res. 2025 | ASKCOS:开源的数据驱动合成规划
今天介绍的是发表在Accounts of Chemical Research上的一项工作——ASKCOS:开源的数据驱动合成规划。近年来,随着机器学习和大规模反应数据集的快速发展,计算机辅助合成规划(CASP)的研究进入了全新阶段。ASKCOS自2016年启动开发,旨在将前沿研究成果转化为免费可用的实用工具,并已在制药、化工以及学术研究中得到广泛应用。该软件集成了逆合成规划、反应条件预测、反应结果预测及路径评估等多项功能,既可通过交互式路径规划器让用户引导探索,也可依托树搜索算法进行自动化设计。其模块化架构覆盖基于模板与无模板的一步模型,并提供溶解度预测与量子化学描述符等扩展能力。凭借易用的界面、开放的代码与持续的社区驱动发展,ASKCOS不仅成为科研人员日常路线设计与决策的助手,也展示了CASP工具在现代化学研究中不断提升的价值与可及性。
Tu, Z.; Choure, S. J.; Fong, M. H.; Roh, J.; Levin, I.; Yu, K.; Joung, J. F.; Morgan, N.; Li, S.-C.; Sun, X.; Lin, H.; Murnin, M.; Liles, J. P.; Struble, T. J.; Fortunato, M. E.; Liu, M.; Green, W. H.; Jensen, K. F.; Coley, C. W. ASKCOS: Open-Source, Data-Driven Synthesis Planning. Acc. Chem. Res. 2025, 58 (11), 1764–1775. https://doi.org/10.1021/acs.accounts.5c00155.
获取详情及资源:
0 摘要
过去十年间,随着机器学习的发展与大规模反应数据集的可用性提升,基于数据驱动的计算机辅助合成规划(CASP)模型取得了快速进展。该综述介绍了自2016年以来持续开发的开源合成规划软件ASKCOS的最新版本中,所整合的多种数据驱动方法与模型。这一工作旨在缩短科研与应用之间的鸿沟,将研究成果转化为免费可用的实用工具。
ASKCOS集成了多种模块:包括逆合成规划、反应条件预测、产物预测,以及若干辅助模块与工具。在逆合成规划方面,软件提供了交互式路径规划器(IPP),支持用户引导式搜索;同时内置树构建器,可结合蒙特卡洛树搜索(MCTS)与Retro*两种经典算法进行自动规划。其核心由四个单步逆合成模型构成,涵盖基于模板与无模板两种策略,能够并行使用、互补优势、提出多样化方案。
在可行性评估上,ASKCOS融合了反应条件推荐、路径打分与聚类、反应结果预测(包括主产物、杂质、位点选择性和区域选择性)等子任务成果。此外,基于以往研究,软件还扩展了溶解度预测与量子化学描述符预测模块,为溶剂适配性及选择性评估提供了更深入的参考。
文章进一步介绍了这些功能如何与其他领域的最新进展相结合,并通过易用的交互界面呈现给化学家。ASKCOS已在日常研究中为数百位药物化学家、合成化学家与工艺化学家提供支持,帮助进行路线构思与决策补充。作者认为,CASP工具正在成为现代化学研究的重要组成部分,其价值与可及性正在不断提升。

图1 | ASKCOS概览 (a) 目标导向的典型合成流程:ASKCOS正是为此设计。目标分子(蓝色圆圈)通过逆合成递归展开,直至得到可购买的起始原料(绿色圆圈)。对于每一步反应,系统都可以预测所需的试剂/条件,并进一步进行可行性评估。(b) ASKCOS的高层次流程图:用户既可以通过图形化界面交互,也可以通过编程接口调用,从而访问不同的预测模块。(c) ASKCOS的模块总结:涵盖多种主题,包括一步逆合成(绿色)、多步搜索(红色)、条件推荐(紫色)、反应结果预测(蓝色),以及各种工具与辅助功能(黑色)。
2 引言
合成规划是指在目标导向合成中选择实验路径与操作方案的一类广义方法。虽然规划合成路线通常需要丰富的化学知识与多年训练,但其定义明确而组合复杂的特性,使这一任务尤其适合算法化推理。形式上,计算机辅助合成规划(CASP)整合了多种计算方法,帮助化学家完成不同任务,包括通过逆合成分析识别可行合成路线、推荐反应条件以及预测反应结果。
自20世纪60年代起,化学家便尝试将有机合成规则编码进自动化计算系统。早期CASP工具依赖专家编码的反应规则与启发式方法,例如在单步逆合成分析中,LHASA系统利用基于分子模式匹配的反应模板,而AIPHOS则首次将逆合成、反应条件推荐与产物预测整合在一个系统中。更近一步的进展如Chematica(现为Synthia),结合现代计算能力与专家整理的规则,可生成复杂分子的合成路线并在实验室中成功实现。随着机器学习的发展和数百万规模反应数据集的可用性,数据驱动的CASP研究重新兴起。大量单步逆合成模型被提出,涵盖基于模板预测或检索、类机器翻译方法以及图编辑预测等。这些模型常与树搜索算法结合,穿越假设反应网络寻找可用的合成路径。机器学习还被应用于反应条件推荐和反应结果预测,类似典型的监督学习场景:模型在历史反应数据上训练,尝试泛化到未见过的目标。
与算法并行发展的,是大量软件工具的出现。专有工具包括LHASA、Synthia、Chemical.ai与IBM的RXN for Chemistry;而开源替代方案包括ASKCOS、AiZynthFinder与Syntheseus。ASKCOS的独特之处在于覆盖范围广泛,不仅聚焦逆合成分析,还尝试解决合成规划中的多个任务。该软件自2016年开始开发,最初使用自动提取的模板与简单树搜索算法(如深度优先与基于启发式的最佳优先)。在过去八年中,它被持续扩展、部署并广泛使用,尤其是在制药与化工企业中得到应用,部分模块甚至被整合进专有设计工具。虽然ASKCOS已被众多研究者验证实用,但此前正式介绍仅限于与机器人流动化学相关的简要报道。
该综述报告ASKCOS的最新版本,综述推动其功能扩展与改进的研究进展(既包括本团队的成果,也包括该领域其他研究)。在核心逆合成功能上,ASKCOS提供两种模式:交互式路径规划器(IPP)与自动化树构建器(Tree Builder),二者均可单独或联合调用多种单步策略,从而融合各自优势。除逆合成外,ASKCOS还为合成规划中的诸多任务提供了解决方案。绝大多数功能都可通过基于网页的图形界面访问,面向药物化学家、合成化学家与工艺化学家,同时其开源与模块化特性(所有预测模块均采用MIT协议发布)进一步提升了易用性。文章其余部分将结合科学原理与实际使用场景,介绍ASKCOS的各项功能(图1)。
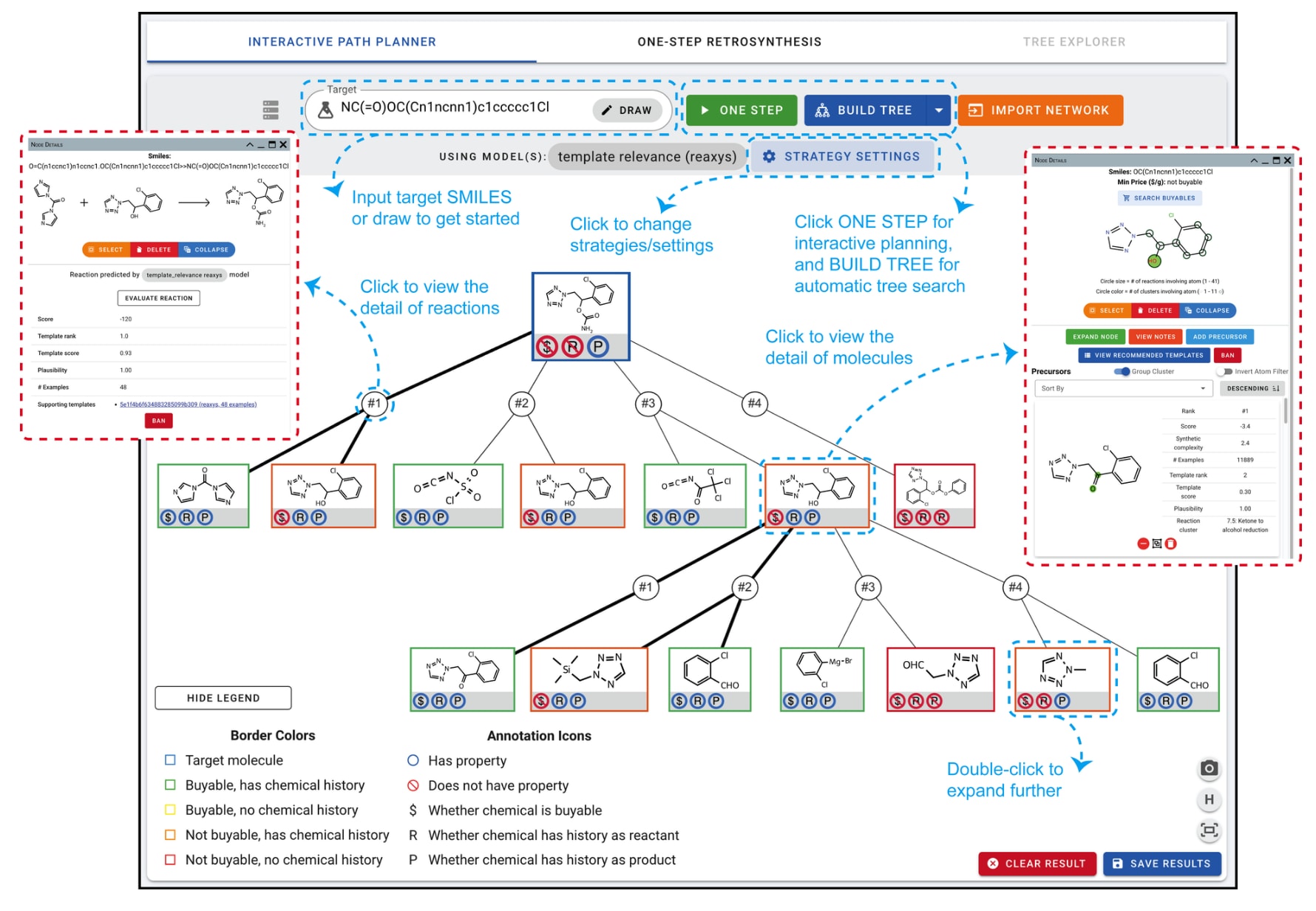
图2 | ASKCOS中交互式规划界面的标注截图
3 数据驱动的逆合成规划
3.1 基于模板相关性模型的交互式规划
自20世纪60年代早期CASP工具出现以来,利用反应模板来建议逆合成断裂一直是流行的做法。ASKCOS中的模板模型借鉴了Segler和Waller提出的神经符号方法,其核心是训练策略网络,对给定目标分子中哪些模板最具策略性与化学合理性进行排序。ASKCOS为这类模型提供了多个检查点,每个模型均基于单一数据源(如Pistachio、CAS Content或Reaxys)的反应模板训练而成。模板的提取与应用由工具RDChiral完成,它能够一致地处理立体化学信息。此外,还基于BKMS数据库的酶反应数据和特定环断裂反应训练了专用模型。由于模板模型基于已发表反应中提取的模板来提出前体分子,其预测结果可追溯到相应的反应先例,因此具有一定可解释性。
图2展示了交互式路径规划器(IPP)中基于Reaxys(2016)训练的模板相关性模型的示例。目标分子可以通过SMILES字符串指定,也可以通过Ketcher绘制分子结构或直接输入常见名称。一键扩展由绿色“ONE STEP”按钮触发,模型给出的若干建议会被添加到画布中,作为目标节点的子节点:圆形表示反应节点,矩形表示分子节点。点击节点会显示更多上下文信息(如图2红色虚线框所示),并可访问额外功能,如屏蔽某个反应/分子(防止其在后续搜索中出现)。反应节点会显示模板评分(即模型返回的概率)、其他评价指标以及可追溯到反应先例的模板详情链接。通过“EVALUATE REACTION”按钮,还可以进一步分析反应,如推荐条件或预测反应结果(详见4.1与4.2)。
分子节点会显示价格,并通过颜色编码标识该分子是否可购买,或是否出现在参考数据库中的已知反应里(见IPP左下角图例)。此外,分子节点提供专属功能,包括展开节点、添加与保存笔记、查看推荐模板等。一旦节点被展开,其详细信息还会列出所有预测的前体分子。用户可以根据不同标准对这些前体进行排序、聚类或按反应中心筛选,并可通过绿色“+”按钮或红色“-”按钮将特定前体添加或移除。
在目标分子完成首次扩展后,用户可自主决定接下来展开哪个分子节点,从而体现了此视图的交互性。例如,可以选择一个不可购买的分子继续展开,直到找到合适的合成路径。若用户希望结合自身思路补充预测结果,还可以通过“ADD PRECURSOR”按钮手动添加前体。探索的网络与笔记可保存到用户个人资料以便后续访问,或导出为JSON文件以供其他化学信息学工具离线处理。
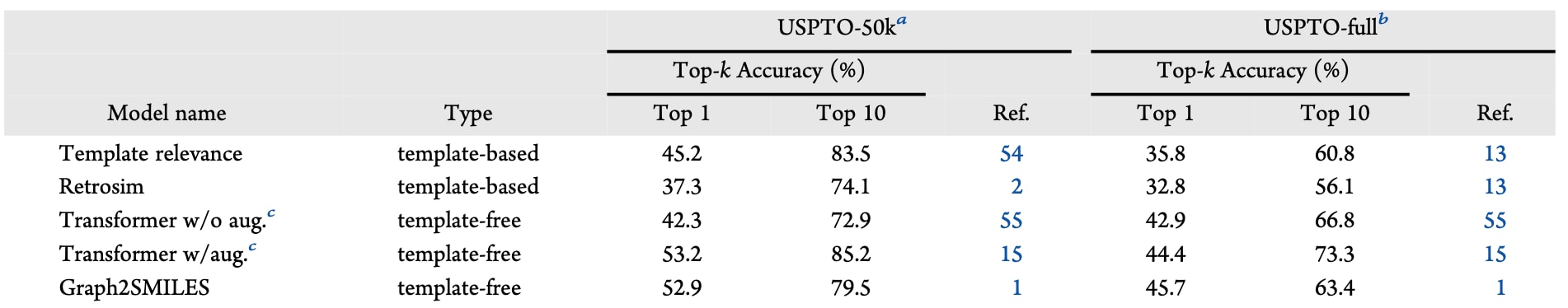
表1 | 以往报道的一步模型在两个常用基准数据集(USPTO50k与USPTO-full)上的Top-k准确率(%)
3.2 基于多模型(含无模板模型)的交互式规划
近年来已报道了大量一步逆合成模型,更全面的总结可见近期的综述。ASKCOS目前整合了四类一步预测策略:前述的模板相关性模型、Retrosim、Transformer与Graph2SMILES。其中,Retrosim是一种基于检索的非学习型方法,通过类比最相似的反应先例来推荐反应;Graph2SMILES则将逆合成建模为图到SMILES的翻译任务,通过图编码器提升了传统SMILES-to-SMILES Transformer的基线性能。
每种模型都有其特定的优势与不足,这些特点并不能完全从公共基准任务(如表1所示)的量化表现中体现,部分原因在于这些基准只覆盖了USPTO中的典型小分子。即便是同一类模型,若训练数据来源不同,其预测结果也可能互补。正因如此,ASKCOS支持整合来自多种策略的推荐结果:当多个模型推荐相同的前体分子时,这可视作对预测更高的置信度。
表2总结了ASKCOS当前可用的一步模型。值得注意的是,基于CAS Content或Reaxys训练的模型在处理更广泛的转化类型时表现更优,因为这些数据集比USPTO与Pistachio等专利数据集更具多样性,并包含更多立体化学信息。ASKCOS在训练时从不混合数据源,而是针对不同数据集分别训练相同模型,从而得到多个检查点,用户既可以单独使用,也可以组合调用。在系统的“STRATEGY SETTINGS”(策略设置)菜单中,用户可自由混搭不同的一步策略,不同策略的查询结果会依次汇总并去重后呈现。每种策略还配有独立设置,例如模板相关性模型的模板数量上限或训练集的选择。在节点详情页面中,来自所有模型的元数据会被聚合,以保证信息简洁。
此外,大多数一步预测策略都支持在新反应数据库上重新训练,例如实验室内部电子笔记系统的专有反应集,从而更好地覆盖不同类型的反应与底物。
表2 | ASKCOS中可用的一步逆合成模型汇总

3.3 基于树构建器的自动多步规划
除了交互式规划(用户逐步选择需要展开的分子节点)之外,自动规划在需要处理大量目标分子时更加便捷。逆合成搜索可通过异步请求同时运行成千上万个目标,代价是用户将失去对扩展方向的直接控制。形式上,自动多步规划可被表述为树搜索或图搜索问题:在搜索的每一次迭代中,系统会选择一个分子进行一步逆合成扩展,并将新的假设反应及其反应物添加到搜索树中。该过程持续进行,直至满足某一终止条件,例如找到所有起始原料均可购买的合成路径。
常见的基线算法包括蒙特卡洛树搜索(MCTS)、证明数搜索(PNS)与A*搜索,这些方法大多受人工智能领域(如AlphaGo)启发。近年来,该领域的研究重点主要集中在改进选择策略,例如利用强化学习、监督学习或化学启发式方法。部分研究还尝试改进扩展策略,将一步模型与多步规划紧密结合。另一些方法则跳出传统树搜索框架,转而采用序列生成或上下文感知搜索。课题组也探索了新的多步搜索表述,例如双向搜索(通过设定特定的基元约束搜索空间)以及高阶逆合成(抽象离去基团的具体身份,转而聚焦更宏观的逆合成策略),但这些方法在撰写该文时尚未整合进ASKCOS。
ASKCOS目前通过*树构建器(Tree Builder)模块支持自动规划,提供两种搜索算法:MCTS与Retro。用户可在“STRATEGY SETTINGS”(策略设置)中进行选择。在IPP画布中指定目标分子后,点击“BUILD TREE”按钮即可启动树构建任务,系统将在后台异步运行。任务完成后会通过弹窗提示结果已可查看。所有结果可在“My Results”页面中找到,条目状态会从“started”变为“completed”。用户既可以在IPP画布中以完整逆合成树形式查看结果(如图3左下角所示的截断示例),也可以在Tree Explorer**中以单条路线的形式浏览(详见4.3)。此外,还可以查看任务使用的设置,并将结果分享给他人(图3右侧示例)。

图3 | ASKCOS中树搜索结果的标注截图
4 数据驱动的合成步骤与路线分析
4.1 反应条件推荐
反应条件预测(包括试剂类别如催化剂、试剂、溶剂,以及温度、当量比等操作条件)是合成规划中不可或缺但常被忽视的一环。条件推荐对任何拟议反应能否在实验中验证至关重要,因为反应结果强烈依赖于具体条件。已有一些反应类型特异性模型,用于预测特定底物家族(如交叉偶联反应)的反应环境,包括温度与压力。与此同时,通用模型也不断涌现,它们不依赖特定反应家族,而是通过多种方法实现,例如多标签分类链、基于SMILES输入的Transformer编码器,或结合文本描述进行的检索增强预测。
ASKCOS集成了一种基于Gao等人方法的数据驱动条件推荐模型,并正在开发第二代版本,进一步支持当量比预测。该模型将条件推荐划分为四个子问题:(1) 试剂身份预测,多标签分类;(2) 温度预测,离散区间分类;(3) 反应物当量比预测,多目标回归;(4) 试剂当量比预测,多目标回归。
图4上方展示了ASKCOS条件推荐页面的截图。与其他模块类似,反应物与产物既可通过SMILES字符串输入,也可通过绘图指定。点击“GET RESULTS”后,模型会生成预测结果,并以表格形式展示多组不同的条件设置。该页面的总体布局与其他模块保持一致:上方为输入面板,下方为预测结果;用户还可以通过蓝色按钮“MODEL”和“SETTINGS”调整所用模型及相关参数。
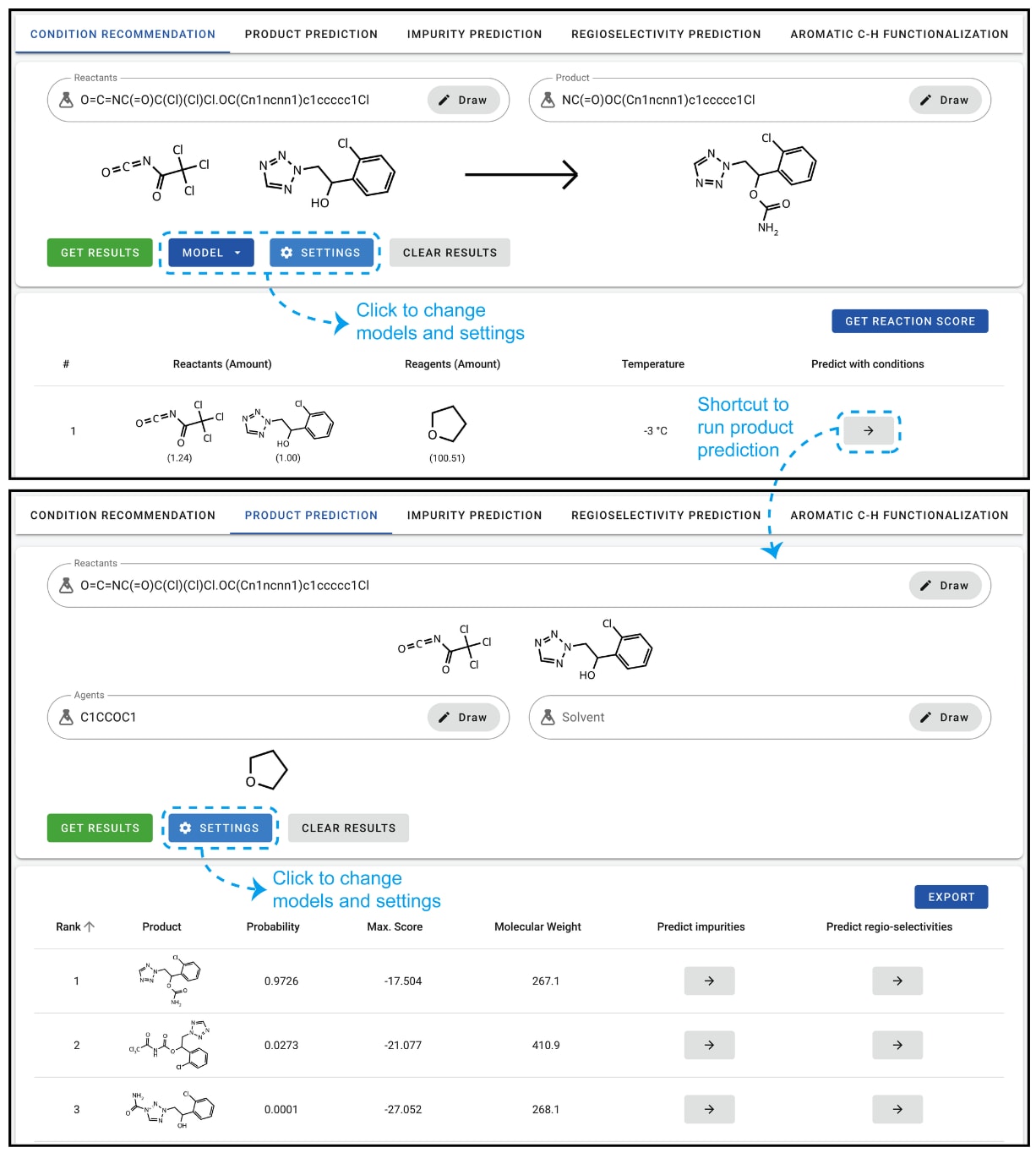
4.2 反应结果预测
除了逆合成之外,合成规划的另一核心环节是反应结果预测。在ASKCOS的工作流程中,该功能主要用于识别化学上不可行或不理想的反应,用户可选择将这些反应从合成树中剔除。许多数据驱动方法将此任务简化为预测主要产物。类似一步逆合成,早期研究多采用模板预测,而后续发展则以基于图编辑、电子流建模及翻译的无模板方法为主。
ASKCOS中集成了多种模型,包括Molecular Transformer的重新实现、课题组开发的Graph2SMILES(与逆合成模型类似,但预测方向相反),以及WLDN5——一种两阶段模型,先预测反应物之间的图编辑,再通过反应物与候选产物进行重排序。
反应结果预测不仅能判断主要产物,还能回答更细致的问题,例如芳香C−H官能化的位点选择性,或在多种区域异构体可能出现的情况下,预测其分布。此外,结果预测还能帮助分析潜在杂质:将杂质视为主要反应的次要产物或副反应产物,可通过考虑低概率预测产物,或通过扩展反应物集合(加入原产物)来模拟过度反应。
图4下方展示了ASKCOS中主要产物预测页面的截图。在该示例中,预测的首要结果为氨基甲酸酯形成,机制为醇对异氰酸酯的亲核攻击(随后发生水解),其概率为0.9726。未水解产物与四唑氮的亲核攻击分别被预测为第二、第三候选,但概率较低,分别为0.0273和0.0001。以往研究表明,这些概率平均而言与预测准确性相关,但并不总能作为单一结果的稳健置信指标。
在同一页面的其他选项卡中,还可访问杂质预测、区域选择性与C−H位点选择性模块。杂质预测模块依赖主要产物预测,并扩展考虑次要产物、过度反应、二聚反应、溶剂加合物以及部分反应物组合。区域选择性与位点选择性的预测细节则见于Guan等人与Struble等人的研究。
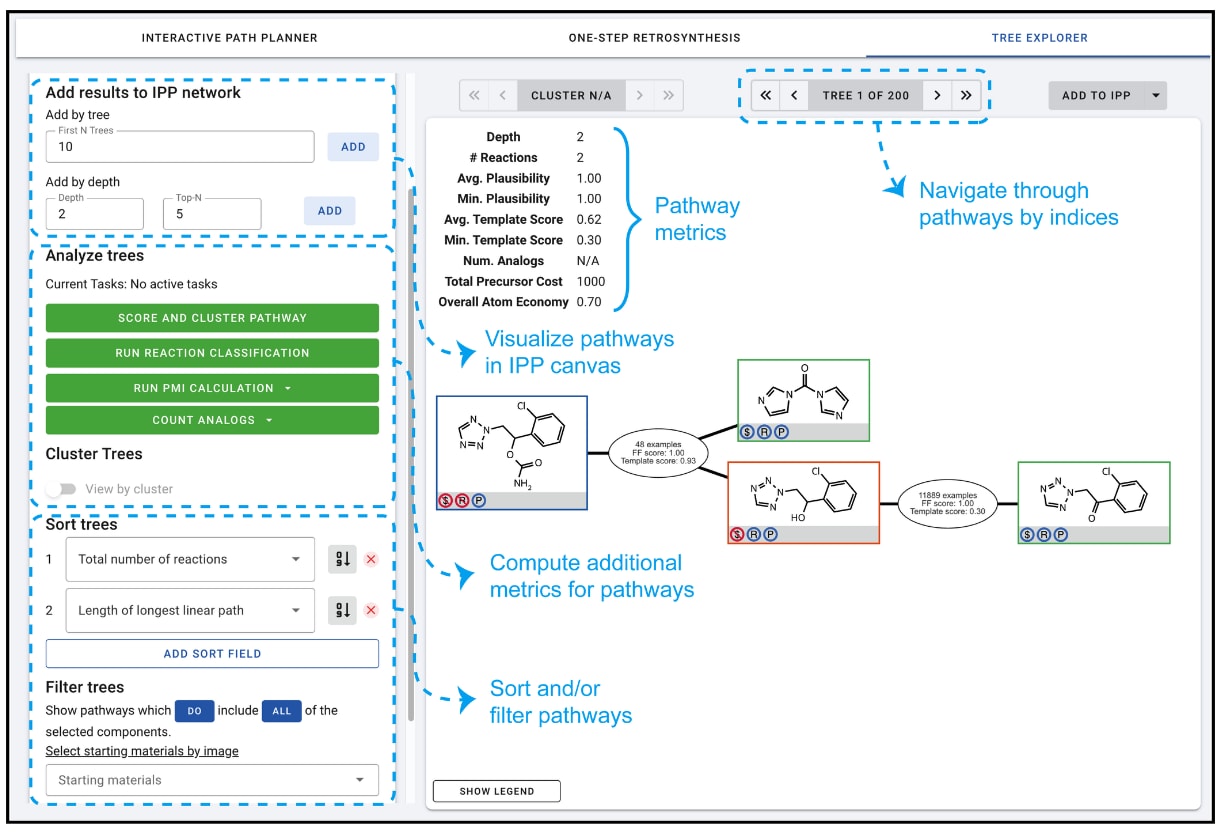
图5 | ASKCOS中树探索(Tree Explorer)程序的标注截图
4.3 路径评分与排序
当为目标分子找到了大量潜在合成路径后,新的挑战随之而来:如何确定最符合化学家目标的路径,而不仅仅是任意一条可行路径。当候选路径数量过大时,人工逐一筛选已不可行。为此,可以先基于训练好的路径嵌入或所涉及的反应类型对路径进行聚类或分组。随后再进行路径层面的评估,以帮助优先挑选更有前景的合成路线。
合成路径的评判标准多种多样,部分指标简单易算,如步骤数、最长线性序列、原子经济性、起始原料成本,或基于基元枚举估算的类似物空间大小(即可多样化程度)。更复杂的指标则可依赖预测模型推导,例如合成人工相似度、总体可行性可能性、基于溶剂使用的绿色化学指标、以及过程物料强度(PMI)。
在ASKCOS中,路径评估主要针对**树构建器(Tree Builder)返回的路径进行,可在结果页面点击“VIEW TREES”进入,见图3。由于上述任一指标都难以单独作为充分标准,ASKCOS在树探索(Tree Explorer)**模块(见图5)中将所有这些指标汇总提供:左侧面板展示操作与选项,右侧画布显示路径及相关指标。在示例中,系统返回了200条路径(默认上限);第一条路径附带自动计算的指标,如路径深度、平均合理性与类似物数量。左侧面板分为三部分:一是可视化多个最佳排序路径并在IPP画布中查看;二是按需计算额外评估指标;三是对路径进行排序或筛选,例如基于特定起始原料进行筛选。
5 数据驱动的辅助预测模型与工具
除了服务于合成规划的“核心”功能外,ASKCOS还包含一系列辅助预测工具与实用模块。其中,研究者开发并集成了两种额外的机器学习模型:溶解度预测模型与基于DFT计算的原子/键级描述符预测模型。
溶解度预测的目标在于提升CASP在工艺化学中的相关性,可用于指导溶剂的选择(反应、液-液萃取或结晶过程)。而ML估算的量子化学(QM)描述符则既可以作为其他预测模型的特征输入,也可单独使用,例如用于人工评估反应的选择性。
此外,ASKCOS还提供其他实用功能,包括基础分子绘制工具、以及基于SMILES的可购买基元搜索。这些功能页面(溶解度预测、溶剂筛选与QM特征预测)统一归类在“Utilities”(工具)选项卡下,并采用与图4相同的标准布局。相关页面截图及更多关于理论原理与实际应用的详细说明则包含在**支持信息(Supporting Information)**中。
6 讨论
作为一个广泛且可扩展的合成规划软件套件,ASKCOS已在MLPDS联盟内外的诸多组织中得到应用。尽管并非所有使用案例都被公开报道,但Struble等人在2020年的综述中总结了部分实例,其中包括ASKCOS如何辅助化学家的工作流程。受到广泛欢迎的功能包括:当自动树搜索失败时的交互式规划模式,以及能够将逆合成建议追溯到文献先例的特性。ASKCOS也为其他工具的发展提供了基础,例如AiZynthFinder中的模板提取策略。Janssen的研发化学家曾在先导优化阶段使用ASKCOS的多个模块,其中一步逆合成API帮助他们将22.2万种醇类化合物筛减至1.57万种可合成候选。Pfizer则利用ASKCOS增强其内部图数据库中的人工创意,Syngenta则将ASKCOS作为多种合成路线生成工具之一进行比较。此外,研究人员也曾利用ASKCOS为候选蛋白酶抑制剂及潜在抗COVID-19药物提出合成路线。
与其他计算工具一样,ASKCOS的所有功能旨在辅助而非取代化学专家。其预测结果受限于训练数据,往往会再现数据中流行的模式与趋势,而不是真正理解其背后的物理化学原理。模型的可解释性差异很大:例如模板驱动的一步逆合成模型可追溯至文献,而基于翻译的模型则更接近黑箱,尽管可能带来创造性,但也可能生成不合理的结果,如在SMILES中错误地增加原子。
作为工具的示范,作者团队进行了合成规划练习(详见支持信息):首先使用树构建器对所有目标进行自动逆合成规划,采用基于Reaxys与Pistachio训练的模板相关性模型,调用默认的可购数据库(几十万种分子),并将搜索树的节点数限制在5000。这样能够为多数目标找到假设性的逆合成路径。随后,进一步利用ASKCOS中的其他模块对那些看似违反直觉或不合理的步骤进行分析,例如交叉验证文献先例或检查排名较低的条件推荐。最后,展示了ASKCOS在自动规划失败时的灵活性,既提供多种树搜索选项,更重要的是提供了用户友好的界面来直接修改拟议路径。
合成规划工具往往生成从高风险到高置信度的各种建议,如何在创造性与保守性之间取得平衡是主观判断的问题。交叉参考文献,无论是否来自ASKCOS,通常都有助于提高路径可信度。一个CASP工具的价值不仅取决于其功能模块,也取决于其在组织内部的部署与定制方式。ASKCOS的开源特性允许本地部署,包括在防火墙内运行以保护专有数据。其部署流程已实现全自动化,仅需复制、粘贴并运行五条命令即可完成;同时,部署也支持定制,可根据需求启用或禁用特定模块,以节约计算资源。用户还可自行重新训练模型或替换默认的基元数据库,这些内容在支持信息中有详细描述。
目前ASKCOS团队会定期基于新的、更优的数据集对模型进行再训练,但其再训练流程也对外开放,用户可在自定义数据集或不同的数据划分(如CAS Content、Pistachio、Reaxys)上训练模型。ASKCOS的发展离不开合作者、同事及社区的反馈。在2023年底进行了一次重大后端重构,引入微服务架构并模块化功能,从而为社区驱动开发奠定基础。此举不仅简化了新模型的集成,也方便高级用户扩展或替换ASKCOS模块。未来将持续增加新模块,包括即将完成的改进版溶解度预测与定量条件推荐,同时优化现有功能的效率与性能。
社区的功能需求更多集中在软件层面(如支持Mac系统、单点登录等),团队将根据优先级推进。研究者认为,CASP乃至更广义的计算机辅助化学,是现代化学研究的重要组成部分,应保持前竞争性与开放性。
在ASKCOS开发之初,可用的开源解决方案非常有限,即使如今多数工具依然是商业化的,即便其方法已在文献中公开。ASKCOS的优势不依赖单一模型或数据集,其数据驱动特性意味着随着新数据的积累与更新,性能能够不断提升。凭借其易用性、可部署性、可定制性与可扩展性,ASKCOS有望成为商业解决方案之外易获取但互补的选择,推动CASP的普及与可持续发展。