J. Med. Chem. 2025 | 肽–药物偶联物治疗效能增强的最新进展
- 最新的PDC创新主要聚焦于提升治疗效果的策略。
- 这些进展涵盖了增强细胞靶向性、提高循环稳定性以及突破生物学屏障的方法。
- 同时,研究者还总结了通过偶联新型功能性药物载荷来进一步增强PDC生物活性的最新探索。

Ma, J.; Wang, X.; Hu, Y.; Ma, J.; Ma, Y.; Chen, H.; Han, Z. Recent Advances in Augmenting the Therapeutic Efficacy of Peptide–Drug Conjugates. J. Med. Chem. 2025, 68 (9), 9037–9056. https://doi.org/10.1021/acs.jmedchem.5c00007.
1 引言
疾病的治疗方式已经从传统的小分子药物逐渐发展到抗体-药物偶联物(ADCs)与肽-药物偶联物(PDCs)。这些药物类型各自具有独特的优势与局限性。小分子药物具有高度稳定性,能够轻易穿透细胞膜,并且可以口服给药,因此成为全球范围内应用最广的药物。但由于难以区分正常细胞与病变细胞,常常导致高毒性与有限的治疗效果。单克隆抗体(mAbs)因其高靶向性与亲和力而被广泛用于炎症、自身免疫疾病与癌症的治疗。通过与小分子药物载荷结合,ADCs能够选择性地将药物递送至病变细胞,显著提升治疗效果。然而,ADCs也存在一些限制,如血液中循环时间过长导致的非特异性毒性、实体瘤穿透性差、研发成本高、药物过早释放以及潜在免疫原性。此外,抗体中大量赖氨酸残基常导致产品异质性,需要位点特异性的偶联技术来获得均一的ADC。实体瘤中致密的结缔组织与细胞外基质进一步阻碍药物的有效渗透,从而削弱疗效。这种独特的肿瘤微环境凸显了开发新型药物与策略的迫切需求。
PDCs的出现为解决ADCs局限性提供了新的可能,同时保留了高靶向性与亲和力。PDCs在结构上与ADCs类似,由肽、连接子和活性载荷组成。但与ADCs相比,PDCs具有分子量小、渗透性强、合成成本低、免疫原性极低等优势。它们已被证明是继ADCs之后在耐药感染和复杂疾病(如自身免疫病、代谢紊乱及癌症)中的一种高效治疗方式。小分子尺寸使其能快速分布至组织并深入肿瘤,促进药物载荷的快速递送。目前已有两种PDC药物获得美国FDA批准。2018年获批的177Lu-DOTA-TATE(Lutathera)是首个用于治疗生长抑素受体阳性的胃肠胰神经内分泌肿瘤(GEP-NETs)的PDC。III期临床试验显示,Lutathera联合长效奥曲肽作为一线疗法,可显著延长Ⅱ级或Ⅲ级晚期GEP-NET患者的中位无进展生存期(PFS),增加约14个月。2022年3月,Pluvicto获批用于治疗PSMA阳性的转移性去势抵抗性前列腺癌。该药物将PSMA特异性的拟肽与放射性核素偶联,能够选择性递送电离辐射至肿瘤细胞。
除肿瘤外,PDCs在其他疾病中同样展现出疗效。例如,多种PDC被设计用于治疗病毒与细菌感染。一种新型PDC由肾脏靶向肽G3-C12(ANTPCGPYTHDCPVKR)、可切割的二硫键连接子与卡托普利载荷组成,在慢性肾病模型中表现出显著的血管紧张素转化酶抑制活性。
PDCs的主要作用机制依赖**细胞靶向肽(CTPs)与膜受体结合,介导内吞或内化过程,使PDC进入细胞。小分子载荷也可通过受体结合促进内吞。内吞是一个依赖能量的过程,需要ATP消耗与囊泡形成来转运PDC。研究表明至少存在四种内吞途径:大吞饮作用、网格蛋白依赖途径、洞蛋白依赖途径以及网格蛋白/洞蛋白非依赖途径。此外,部分PDC中的细胞穿透肽(CPPs)**能够通过能量非依赖机制直接跨膜,改变接触点的膜动力学,促进PDC进入细胞。一旦进入细胞,PDCs被转运至内体或溶酶体,在低酸性与高酶浓度的环境下,肽与载荷被释放至细胞质、细胞核或其他细胞器,从而发挥其功能。
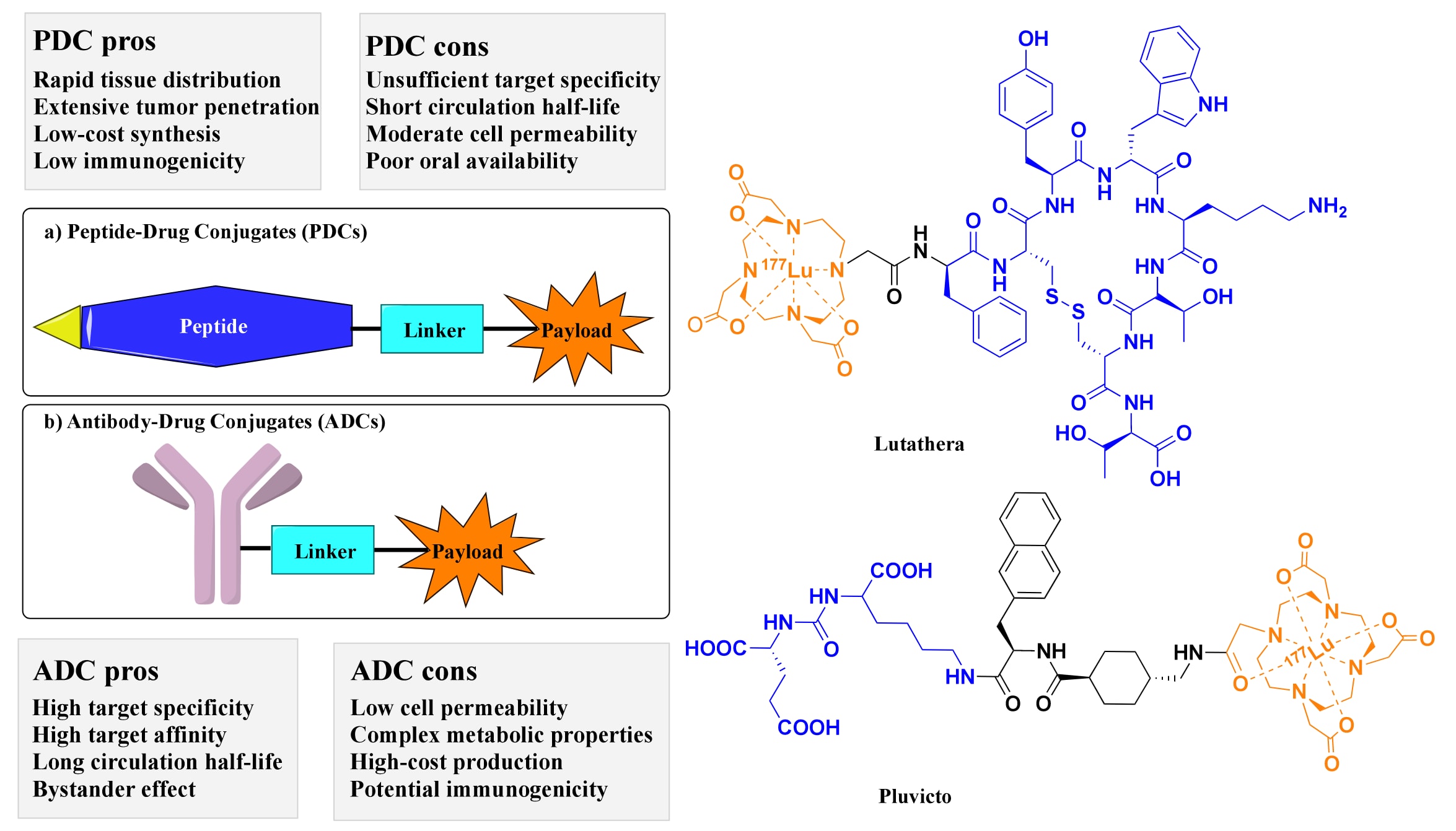
图1 | 肽-药物偶联物(PDC)的示意结构及已获FDA批准的PDC的化学结构。 (1) Lutathera包含一种来源于含酪氨酸的生长抑素类似物Tyr³-octreotate (TATE)的细胞靶向肽(CTP),通过酰胺键连接子与载荷结合,该载荷由大环螯合剂四氮杂环十二烷四乙酸(DOTA)与β发射放射性核素镥-177 (¹⁷⁷Lu)结合而成。(2) Pluvicto包含一种带有PSMA结合基序Glu−NH−CO−NH−Lys的CTP,连接子由2-萘基-L-丙氨酸与氨甲环酸构成,载荷部分与Lutathera相同。(3) 图中蓝色代表CTP,橙色代表载荷,黑色代表连接子。
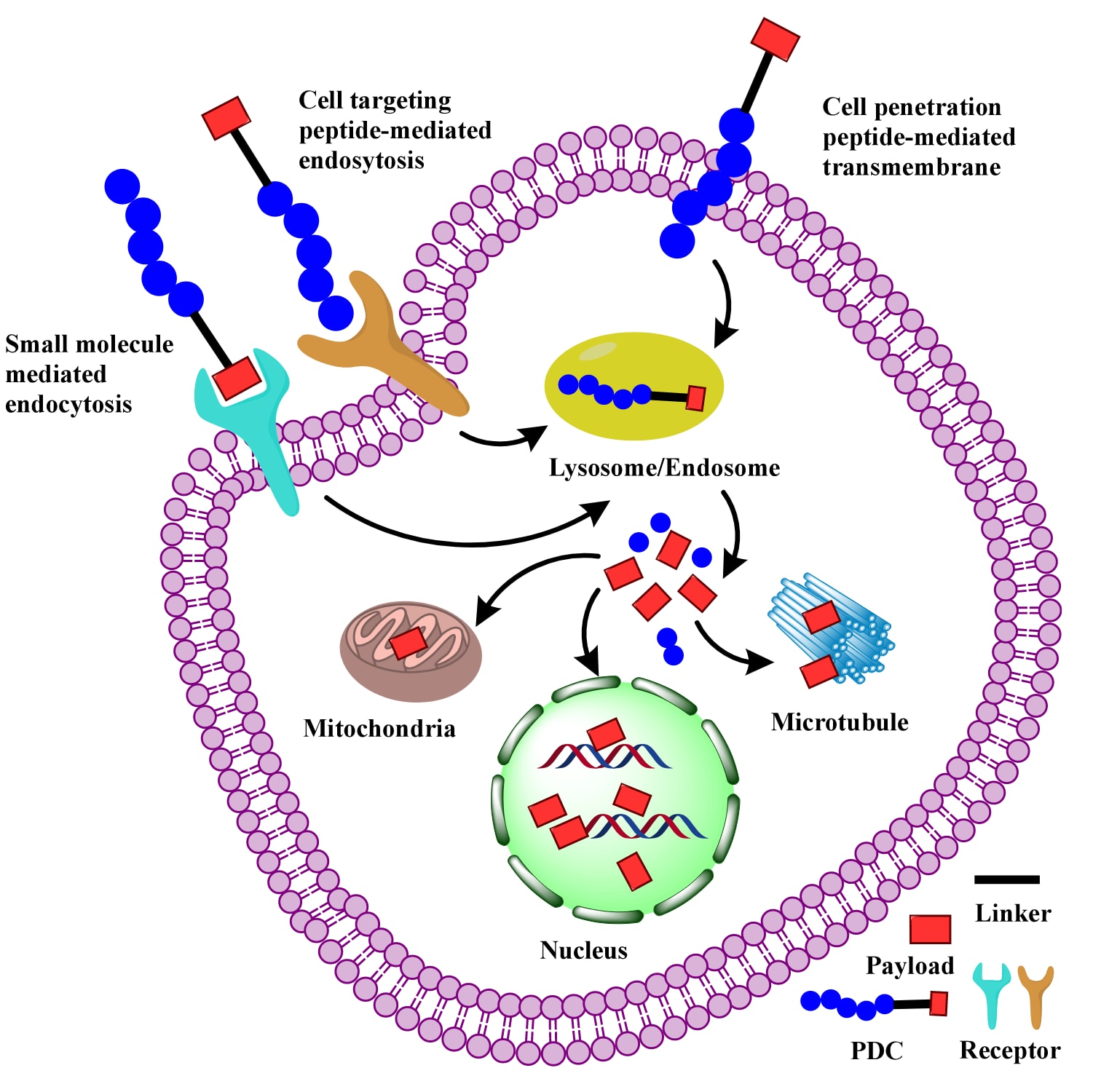
图2 | PDC的作用机制。 PDC可通过CTPs或小分子载荷介导的内吞进入细胞,亦可通过CPPs跨膜。进入内体或溶酶体后,在低酸性和高酶浓度的环境下,载荷被释放至细胞质、细胞核或其他细胞器中发挥功能。
2 PDC的组成与偶联策略
在PDC的设计中,首先需要确定针对特定靶点或疾病的肽段或小分子载荷,这是逻辑上的起点。然而,如何高效地将载荷递送至靶点部位始终是一项具有挑战性的任务。PDC中的肽部分主要负责靶向特异性,从而实现位点特异性的载荷递送,并降低毒性。过去十年中,PDC肽段的创新(Figure 3),例如新型细胞靶向肽(CTPs)、细胞穿透肽(CPPs)、自组装肽(SAPs)以及刺激响应肽(SRPs),显著提升了药物载荷的治疗效果。多数肽类药物源自激素模拟物或天然产物。然而,如何设计具有高效性与选择性的肽段来靶向受体的配体结合结构域依然困难。为此,研究者发展了多种编码与展示技术以筛选高效肽序列,包括噬菌体展示、肽蛋白分子内拼接、mRNA展示,以及“一珠一肽”或分裂-汇聚策略。近年来,核苷酸编码的大规模文库筛选技术推动了非天然基团与非蛋白源氨基酸在难以成药靶点上的应用。通过这些方法筛选出的肽段通常对靶蛋白具有高亲和力。
连接子在PDC设计中同样至关重要,因为它直接影响PDC的安全性与效力。目前主要有两类连接子(Figure 4):可切割型与不可切割型。前者常对pH、谷胱甘肽或蛋白酶(如二肽连接子中的组织蛋白酶B)敏感;后者如噻唑、肟或硫醚类,则能增强PDC在人体内的循环稳定性。
偶联步骤是PDC合成中的关键环节。传统的酰胺或酯键形成已被证明是连接肽段与载荷或连接子的高效手段。近年来,点击化学反应因其高效、快速与温和条件而受到广泛关注(Figure 5)。其中,叠氮-炔烃Huisgen环加成反应最为常用,能在水相或有机溶剂中生成三氮唑单元,被广泛应用于ADC与PDC,尤其是多聚PDC的合成。然而,该反应需要金属催化剂,存在安全隐患,因此发展出了应变促进的叠氮-炔烃环加成(SPAAC),无需金属催化剂即可进行。但SPAAC反应活性过高,可能导致区域选择性与化学选择性较差。另一种替代方法是Staudinger连接反应,它能在水相中通过膦化合物与叠氮反应生成酰胺键,无需金属催化剂。传统的伯/仲胺与羧酸偶联也可形成酰胺。
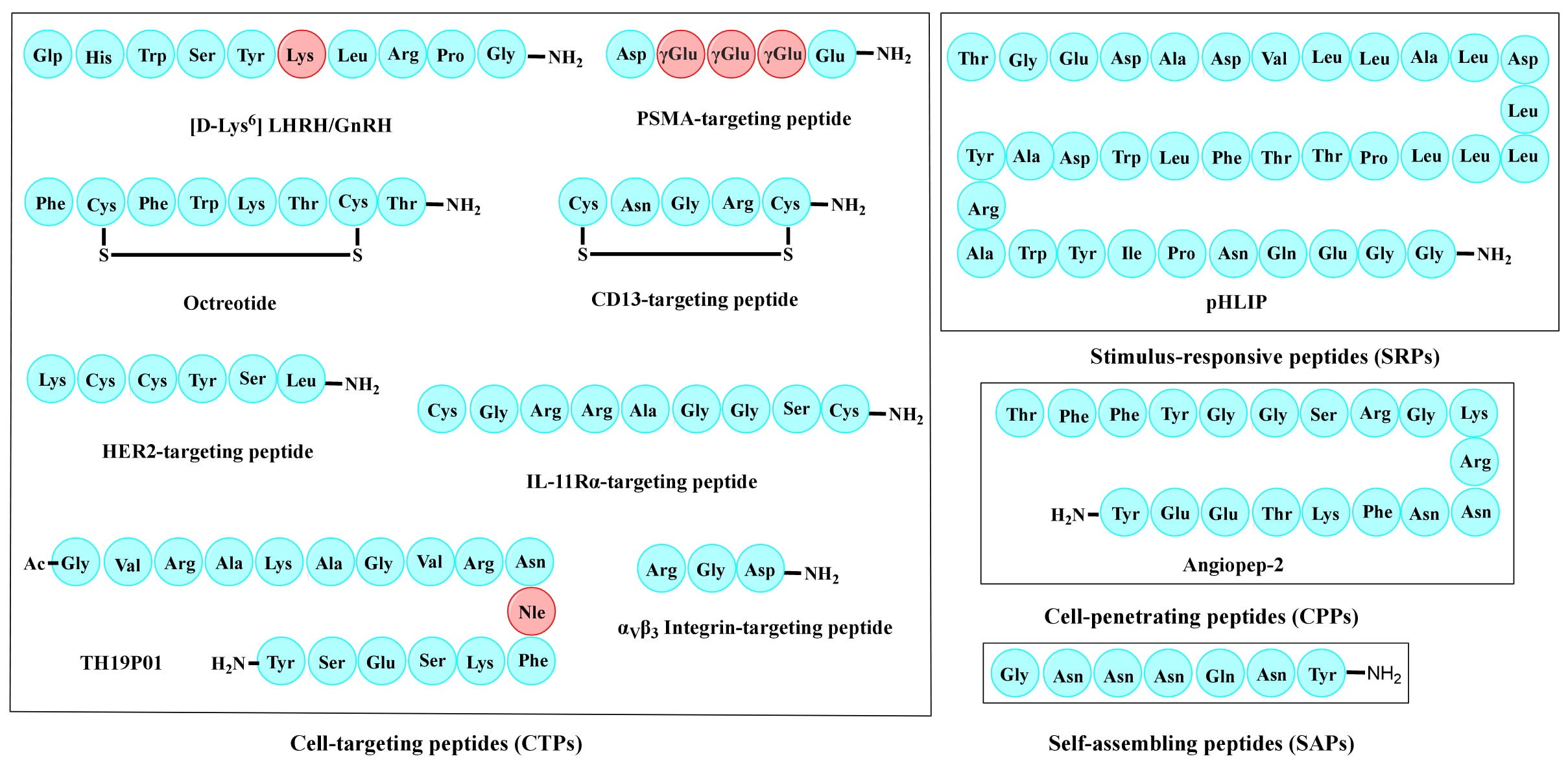
图3 | PDC中常用的代表性肽。 左侧方框展示了一些典型CTP的氨基酸序列,其中红色氨基酸为非天然氨基酸。右侧方框分别展示了SRP、CPP与SAP的实例。
在实际应用中,研究者利用多种偶联策略构建新型PDC。例如,一种针对特发性肺纤维化的PDC采用叠氮-炔烃点击反应,将识别αVβ6整合素的小环肽c(AmpLRGDL)与酪氨酸激酶抑制剂nintedanib偶联合成。另一例是EphA2靶向型PDC,通过将EphA2结合肽与含叠氮基的己酰紫杉醇(PTX)片段偶联而成。肽段序列中常含半胱氨酸,可用于巯基-烯烃的加成反应(Michael加成),该反应通常要求烯烃连接电子吸引基团,并在水相与有机相中均有效。另一类偶联方式是羰基与氧肟基缩合生成肟,或与肼生成腙。酯键与氨基甲酸酯也常用于PDC合成。例如,研究者通过酯键连接将疏水性药物PTX与CPP偶联,连接子采用琥珀酸,使其成为一种pH可切割连接子 。当PDC进入癌细胞后,酸性环境会水解酯键,从而释放PTX载荷。
总体而言,PDC的合成充满挑战,特别是在终端偶联化学选择上,需要根据PDC的整体结构在安全性、效率与反应条件等方面进行全面权衡。本文不再赘述PDC的各组成部分,因为已有多篇高水平综述系统总结了肽段、连接子与载荷的相关研究。接下来,将重点介绍正在研发中的癌症靶向型PDC,它们在临床试验中的应用,以及近期提升PDC治疗效果的新策略。
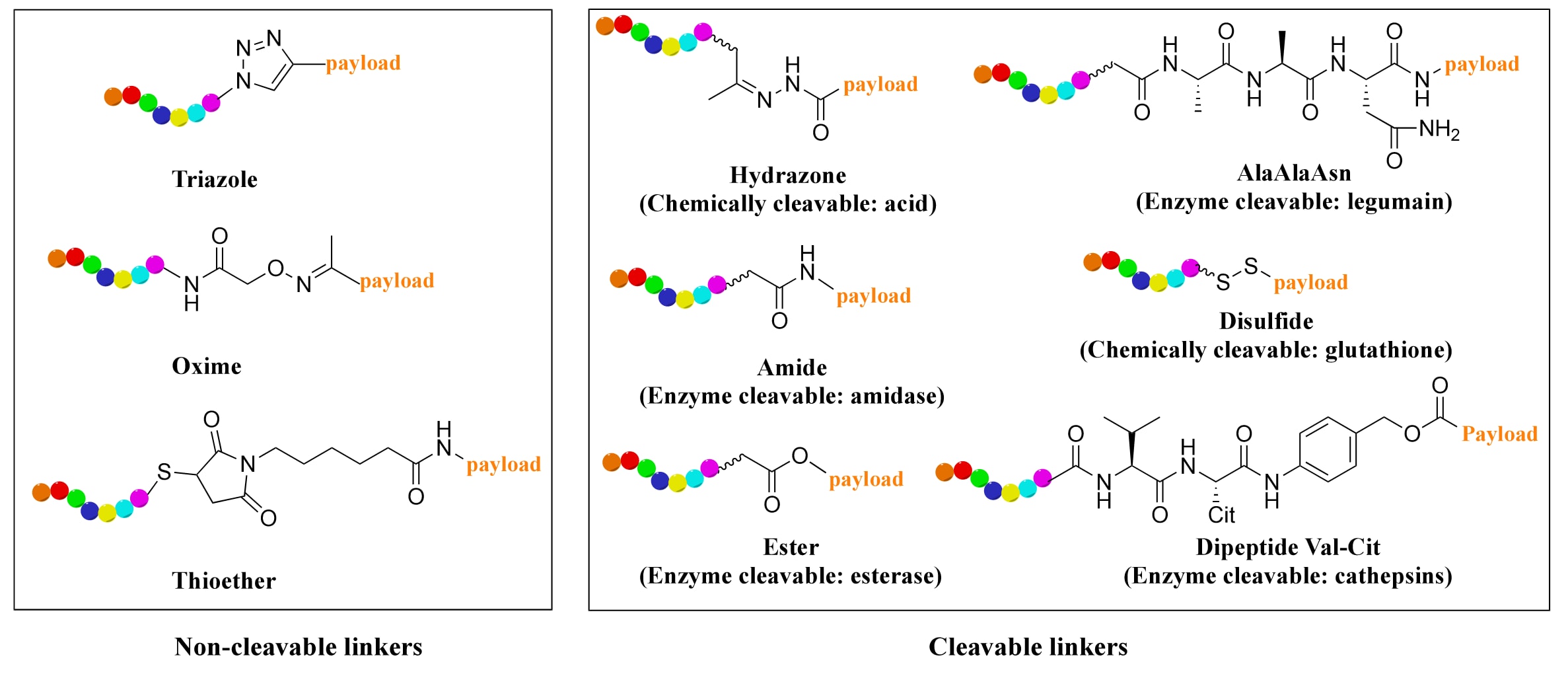
图4 | PDC中不同连接子的化学结构。 肽链以彩虹色表示,连接子以黑色表示。
3 提升PDC治疗效果的策略
PDC若要发挥更大治疗潜力,必须在细胞靶向能力、循环稳定性与渗透性方面得到提升。然而,目前仅有极少数PDC能够通过口服给药并在体内达到足够的系统浓度。因此,高效的细胞内运输与靶点递送是克服现有不足的关键。为此,研究者们开发了多种策略,以改善靶向能力、循环稳定性、毒性管理以及血脑屏障(BBB)的穿透性,从而优化PDC的药代动力学特性与疗效(Figure 6)。
3.1 改善PDC的细胞靶向性
3.1.1 多价性策略
细胞靶向肽(CTPs)具有中等靶向特异性与亲和力,能够将药物载荷高效递送至过表达特定靶点的细胞。为增强CTPs的靶向性与结合力,研究者发展了二聚体或三聚体肽段策略,从而提升治疗效果(Figure 6)。例如,RGD肽以其靶向整合素αVβ3而广泛用于肿瘤治疗。研究表明,二聚体RGD肽的肿瘤亲和力比单体提高10倍,且在肿瘤中保留时间更长;三聚体或四聚体衍生物的活性优于单体与高度聚集的多拷贝形式。类似地,二聚体或四聚体cRGD−PTX偶联物相比PTX表现出更强的靶向结合力。
在其他靶点中,EphA2受体在多种实体瘤中过度表达,与癌症发生、血管生成及转移密切相关。二聚体EphA2结合肽相比单体亲和力提升约13倍,并在乳腺癌与胰腺癌模型中有效抑制转移。类似地,二聚体环肽与光敏剂吲哚菁绿偶联后,在乳腺癌的光热治疗中效果显著优于单体。针对HER2的二聚体环肽PDC亦展现出增强的结合亲和力、抗肿瘤活性以及更低的系统毒性。这些结果强调了多价性策略在增强PDC细胞靶向性方面的重要性。
3.1.2 双特异性靶向
双特异性策略旨在同时作用于同一蛋白的不同结构域或不同靶点。例如,一种新型PDC结合了EGFR胞外靶向肽(LARLLT)与EGFR胞内靶向载荷吉非替尼,在肺癌细胞中表现出优于单药的抗肿瘤效果。另一种双特异性融合肽可同时结合HER2的II区与IV区,偶联喜树碱后在HER2阳性乳腺癌小鼠模型中显著增强抗癌活性。
3.1.3 肽段的卤代修饰
卤代修饰是调控肽类药理活性的新兴手段。通过在特定位点引入氟、氯、溴或碘,可以改善药物的药理性质与生物活性。例如,在RGD肽中引入卤代色氨酸增强了对αVβ3整合素的亲和力与选择性;含氯的cryptophycin衍生物对宫颈癌细胞的活性提高了8倍;溴代抗菌肽活性提升2倍,同时对正常细胞的毒性降低。这表明卤代修饰有助于提高PDC的靶向性与疗效,值得进一步研究。
3.1.4 自组装肽(SAPs)、刺激响应肽(SRPs)与纳米结构形成
当PDC由亲水性肽段与疏水性药物组成时,可自组装成纳米管、纳米纤维、水凝胶或超分子丝等多种纳米结构。短肽与小分子疏水单元的偶联能够赋予新的自组装特性。氨基酸的类型与序列决定了其二级结构(如螺旋、β-折叠或无序构象),进而影响自组装能力。SRPs则能够响应pH、温度、氧化还原、电光或酶的刺激。其中,pH响应性肽在肿瘤靶向中尤为突出,例如pHLIP肽在低pH的肿瘤微环境中形成α螺旋并插入膜内,将药物递送入细胞。基于pHLIP的PDC CBX-12已进入临床II期,用于治疗铂耐药或难治性卵巢癌。
此外,结合RGD靶向肽、自组装肽(GNNNQNY)与喜树碱的PDC(SAP-CPT)能在肿瘤内原位自组装成纳米结构,增强内吞并提升肿瘤杀伤作用。另一种ROS响应型PDC通过硫缩醛连接子实现线粒体靶向,在肿瘤内释放喜树碱以选择性杀伤癌细胞。还有研究利用两分子阿霉素偶联自组装成纳米胶束,并通过透明质酸(HA)包裹增强对CD44过表达肿瘤的靶向。PEG化的PDC也能形成纳米颗粒,利用**增强渗透与滞留效应(EPR)**被动富集于肿瘤部位。
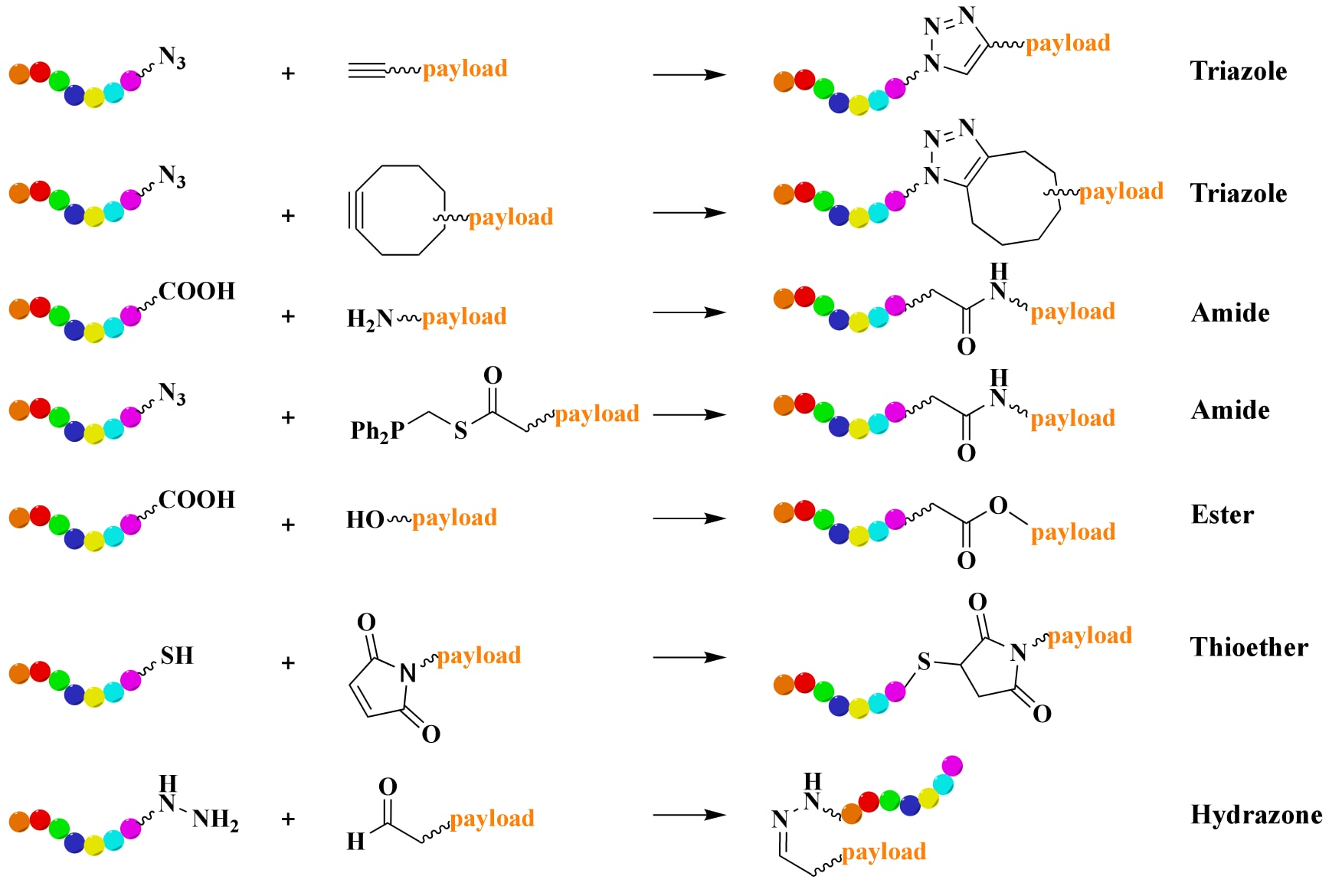
图5 | PDC常见偶联反应的示意图。 肽片段以彩虹色高亮显示。
3.2 通过细胞穿透肽(CPPs)突破生物学屏障
如何让药物穿越血脑屏障(BBB)或多层细胞构成的上皮屏障,是PDC研发中的重大挑战。CPPs通常由5–30个氨基酸组成,能够在无受体参与的情况下跨膜进入细胞,常通过内吞作用携带药物、核酸或探针跨越屏障。CPPs可按理化性质分为阳离子型、疏水型或两亲型。早期的代表如HIV-1转录激活蛋白(TAT)与RGD序列已证明其递送多种分子载荷的能力。精氨酸丰富的CPP进入细胞的效率高于赖氨酸多肽,但短于六残基的聚精氨酸则无效。不过,这类肽段在动物实验中表现出一定肾毒性,因此仍需优化。近年来,环化CPP成为新趋势,可降低结合膜所需的能量并提升渗透性。例如,环状亲水肽EPP6在无正电荷条件下仍表现出优异的跨细胞转运能力;肽-铋双环结构则使细胞摄取量提升了一个数量级。此外,环化的阳离子CPP在抗菌活性方面也优于线性肽。尽管CPP在递送上具有优势,但普遍缺乏细胞特异性。为解决这一问题,研究者发展了可激活型CPP,即通过可切割连接子掩盖其穿膜功能,待到肿瘤组织中被蛋白酶水解后才激活。基于此策略的多柔比星(DOX)-CPP前药在肿瘤模型中展现出显著疗效。
在中枢神经系统药物递送中,BBB是最重要的障碍。其跨膜机制包括被动扩散、载体介导、受体介导、吸附介导及细胞介导的转胞作用。BBB特异性受体,如VEGFR、EGFR、转铁蛋白受体(TfR)、整合素受体和低密度脂蛋白受体,成为CPP设计的切入点。Angiopep家族源自人胰蛋白酶抑制剂的Kunitz结构域,其中Angiopep-2能与LRP1受体结合并促进脑内转运。基于此的PDC ANG1005由三分子紫杉醇(PTX)通过琥珀酸连接子偶联至Angiopep-2,能通过LRP1介导的转胞作用穿越BBB并在溶酶体中释放PTX。I/II期临床试验显示,ANG1005在恶性胶质瘤及乳腺癌脑转移患者中展现出延长生存的潜力,目前III期试验正在进行。另一种新型PDC利用TfR靶向肽(HAIYPRH)与载荷SN-38结合,成功实现了跨BBB递送并在胶质母细胞瘤中诱导细胞毒性。
3.3 CPP与CTP的联合策略
CPP具备高效的细胞内转运能力,但缺乏靶向性;而CTP具备特异性,却难以高效跨膜。因此,将二者结合成为提升PDC递送效率的有效方案(Figure 6)。例如,将RGD肽与精氨酸残基结合的多糖衍生物能够实现协同靶向并增强癌细胞的内吞,同时降低对正常细胞的毒性。进一步的研究表明,结合整合素靶向肽(RGD)、CPP与阿霉素或紫杉类药物的PDC能够显著提高抗肿瘤活性。其机制在于CPP促进细胞摄取,而CTP将药物导向特定受体阳性细胞。在前列腺癌与乳腺癌模型中,基于RGDC靶向肽、环状CPP与cabazitaxel构建的PDC通过谷胱甘肽切割与酯键水解释放载荷,展现出增强的抗癌效果。由此可见,CPP与CTP的结合具有显著的协同作用,是提升PDC靶向性与疗效的有效策略。
3.4 提升PDC的循环稳定性
易被蛋白酶降解与快速肾清除是肽药研发长期面临的难题。PDC因分子量小,更易被水解,导致血浆半衰期短,药物难以在体内维持有效浓度。为此,研究者提出了多种方案,包括环化、白蛋白结合、氨基酸取代与修饰(Figure 6)。一项研究表明,基于抗体辅助的PDC在胶质瘤小鼠模型中延长了循环半衰期并改善了疗效。
3.4.1 环肽、双环肽与Stapled肽
环肽相比线性肽在结构与代谢上更稳定,因其末端被“封闭”而不易被外肽酶切割。例如,环状RGD在pH 7条件下比线性RGD稳定性高30倍。环化不仅提升了稳定性,还增强了受体结合力。基于环化的抗PD-L1肽表现出比线性肽更优异的体内抗肿瘤活性。Stapled肽通过插入脂肪族桥锁定α螺旋构象,从而提升蛋白水解稳定性与膜通透性。基于p53的Stapled肽被证实可增强肽段稳定性与结合力。双环肽则在构象上更加受限,亲和力与抗降解性能显著优于单环肽。例如,靶向nectin-4的双环肽PDC BT8009在临床前展现出强大的抗肿瘤活性,目前正在开展针对多种癌症的临床试验;另一个EphA2靶向型PDC BT5528在I期试验中也显示出良好安全性与抗癌效果。
此外,Knottin肽与多环肽因其二硫键交联结构而具备高热稳定性与蛋白水解稳定性,被认为是潜在的口服给药候选。最新研究设计了一种Knottin肽-药物偶联物(KDC),能够选择性递送吉西他滨至整合素阳性的恶性细胞,并在细胞内释放载荷,显示出优异疗效。
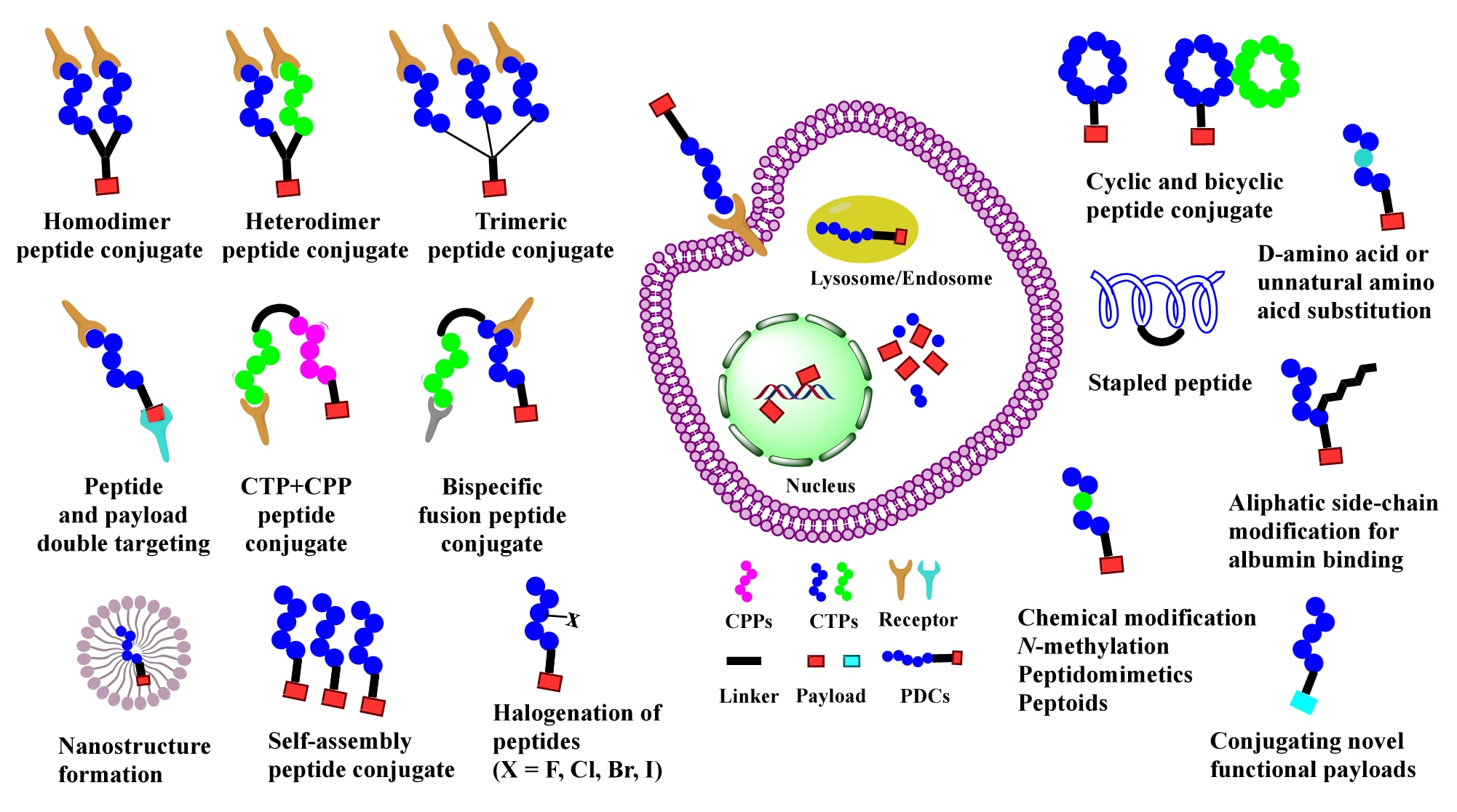
图6 | 当前PDC的挑战与优化策略概览。
3.4.2 白蛋白结合策略
PDC因分子量较低,极易通过肾脏快速清除。为解决这一问题,一种重要策略是通过白蛋白结合来提高分子量并延长其在体内的滞留时间。白蛋白是人体内含量最丰富的血浆蛋白,分布于血液、淋巴及组织间液中,具有维持渗透压及转运外源药物与内源小分子(如胆红素、金属、激素及脂肪酸)的重要作用。人血清白蛋白(HSA)在体内半衰期约19天,这一特性推动了多种白蛋白结合基团的开发(Figure 7)。例如,4-(对碘苯基)丁酸衍生物可与白蛋白形成稳定的非共价结合,显著延长[¹⁷⁷Lu]Lu-DOTA-TATE在血液中的平均滞留时间。大麻二酚亦可高亲和力结合HSA与γ-球蛋白。另一类截短的Evans Blue衍生物(tEB)能够与白蛋白二聚,形成“夹心”结构保护肽免于降解。研究还显示,带有阳离子赖氨酸残基的PDC具有较强的白蛋白结合能力和生物活性。
白蛋白结合策略最早在胰岛素药物中得到验证。通过脂肪酸酰化(Figure 8),研究者实现了长效胰岛素的开发。地特胰岛素(detemir)与德谷胰岛素(degludec)均通过B链赖氨酸连接脂肪酸而显著延长作用时间。新一代的icodec胰岛素结合脂肪二酸衍生物,其血浆半衰期长达196小时,并于2024年在欧洲获批用于2型糖尿病的治疗。类似策略也应用于GLP-1类似物,如CJC-1131和semaglutide,后者通过赖氨酸侧链连接C18脂肪酸延长了循环半衰期。tirzepatide则在结构上结合了C20脂肪酸修饰,实现了更强的稳定性。近期,基于脂肪酸修饰的长效PDC亦被开发,在小鼠肿瘤模型中表现出增强的血浆稳定性与延长的抗肿瘤作用时间。需要注意的是,尽管大分子如PEG、多唾液酸或羟乙基淀粉也可降低肾清除,但其聚合效应可能削弱肿瘤渗透能力,从而影响疗效。最新策略探索了聚合物−药物偶联物,通过作用于肿瘤相关髓系细胞改善免疫治疗反应。
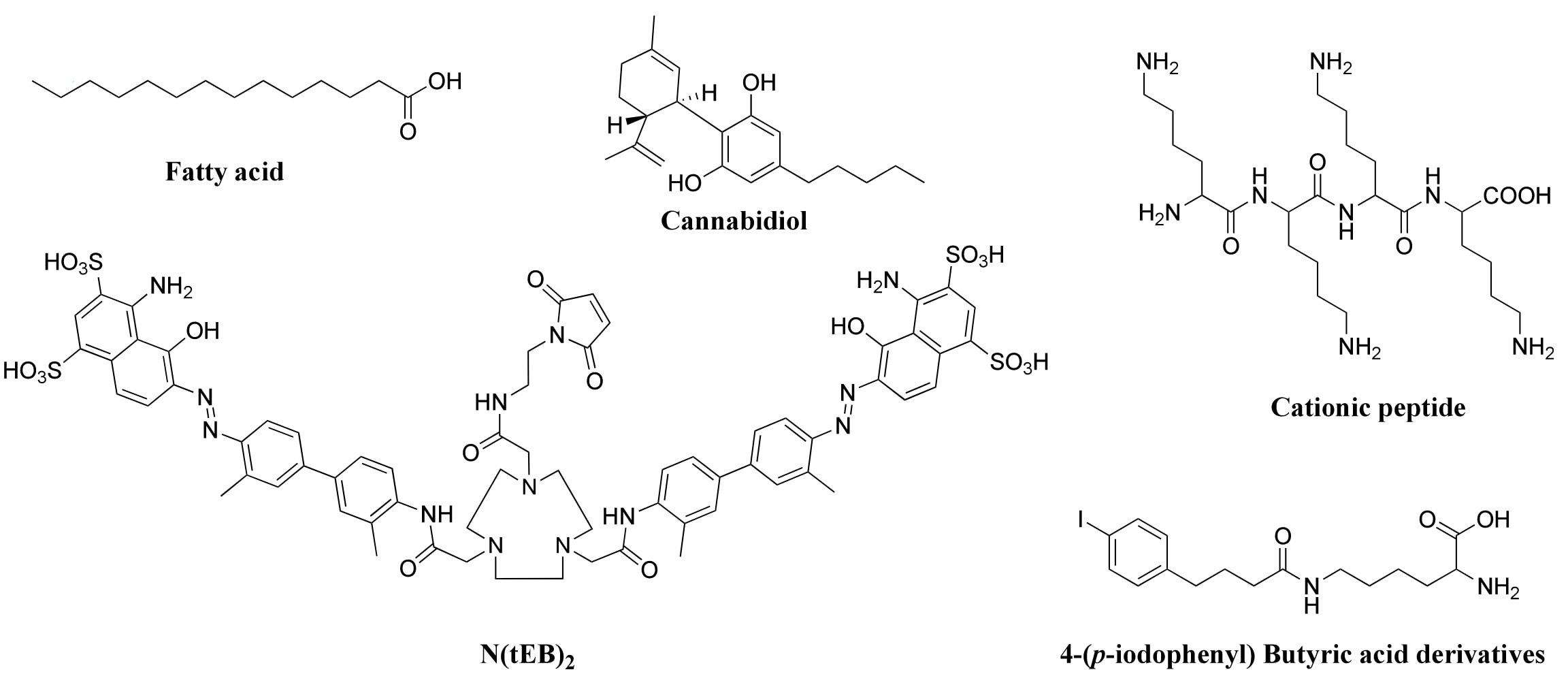
图7 | 小分子白蛋白结合基团的化学结构。
3.4.3 氨基酸取代与化学修饰
为进一步提升肽类的生物活性与稳定性,研究者采用了多种化学修饰手段,包括D-氨基酸或非天然氨基酸替换、N-甲基化以及拟肽(peptidomimetics)与拟肽类(peptoids)的设计。D-氨基酸较L-氨基酸更耐受蛋白酶降解。例如,将L-氨基酸序列部分或全部替换为D-氨基酸的多肽在血清中的稳定性显著提高,同时增强了肿瘤靶向性。值得注意的是,D-氨基酸替换可能因构象改变而影响活性,因此需权衡。另一策略是通过改变等电点与电荷分布调节血浆半衰期。例如,**甘精胰岛素(insulin glargine)**通过单个氨基酸替换及C端精氨酸延长了作用时间。类似地,在GLP-1序列中将Ala8替换为Gly,显著提高了对DPP-IV的抵抗力。
化学修饰亦是重要方向。N-甲基化通过改变氢键网络提高了多肽稳定性;**Aib(α,α-二取代氨基酸)**的引入能稳定α螺旋与β折叠结构,从而延长半衰期。semaglutide结合Aib替换与脂肪酸修饰,使其在人类体内的半衰期达到165小时。拟肽与拟肽类因其耐受蛋白酶降解、降低免疫原性及提高细胞通透性而被广泛研究。例如,临床候选药BMTP-11由IL-11Rα结合肽与诱导凋亡的拟肽组成,在骨肉瘤与前列腺癌模型中展现出显著疗效。拟肽−肽水凝胶也被证明在长效药物递送系统中具有潜力。
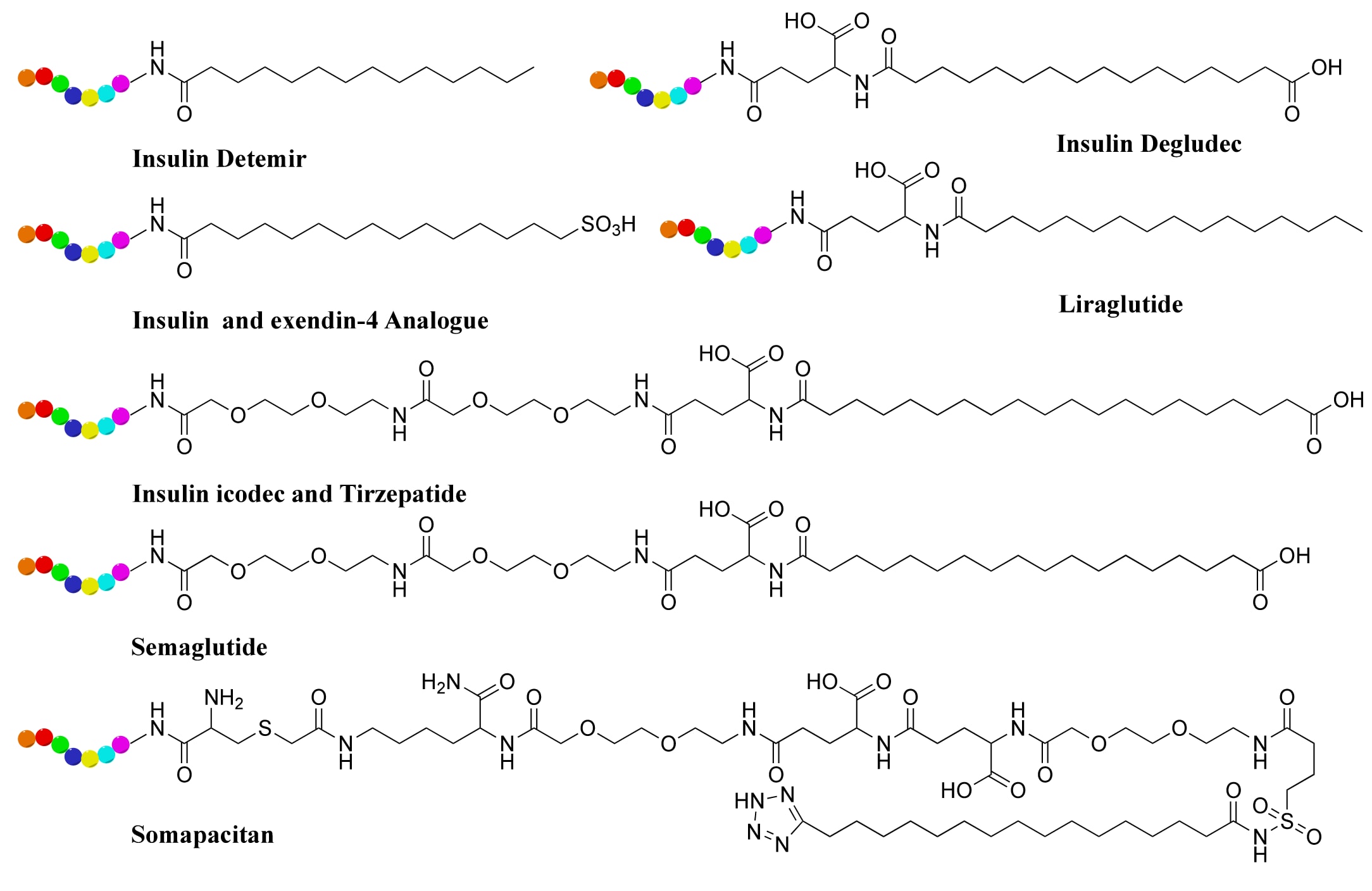
图8 | 用于长效肽药物与PDC的白蛋白结合脂肪酸(以黑色表示)的化学结构。
3.5 偶联新型功能性载荷
由于生物分布、细胞摄取率不足及循环中的偶联物失活,只有少部分PDC载荷能到达细胞内靶点。因此,载荷必须具备足够的效力,即便在低浓度下也能有效杀伤靶细胞。PDC载荷往往比传统化疗药物更具毒性。与ADCs相比,PDC因肽分子量小、渗透性好,可支持更高
传统PDC载荷包括微管靶向药物(如紫杉醇PTX)、抗有丝分裂剂(MMAE、DM1)、DNA损伤类药物(如吡咯并二氮杂卓二聚体)及拓扑异构酶I抑制剂(SN-38)。近年来,研究拓展至细胞因子、溶解性肽、毒性蛋白、蛋白降解靶向嵌合体(PROTACs)、寡核苷酸及PD-L1降解剂等新型载荷。PD-L1作为癌细胞逃避免疫监视的重要检查点受体,降解其表达可增强免疫治疗效果。例如,BMS-L1-RGD通过cRGD偶联PD-L1降解剂BMS-8,在黑色素瘤模型中有效抑制肿瘤且副作用低。另一款新型PDC PMT-O9-1A,由抗PD-L1肽与降解剂palmatine通过二硫键连接而成,可双重抑制PD-L1(阻断与降解),在肺癌模型中显著提升疗效。
溶解性肽是另一类创新载荷,通常由15个氨基酸组成,富含亮氨酸与赖氨酸,通过破坏细胞膜磷脂双层杀死癌细胞。由于其非特异毒性,研究者开发了多种靶向结合策略。例如,将溶解肽与EGFR靶向肽或IL-4R靶向肽通过甘氨酸连接偶联,显著提升抗肿瘤活性。QR-KLU则结合VEGFR靶向肽与溶解肽,表现出强效的肿瘤抑制作用,并增强了抗PD-1治疗的疗效。在临床中,EP-100由激素基肽与阳离子α螺旋溶解肽ClIP-71组成,能通过膜溶解作用杀伤LHRH受体阳性肿瘤细胞。虽然其与PTX联合治疗卵巢癌的客观缓解率与单用PTX相当,但部分肝转移患者获益明显,且研究发现EP-100可上调PD-L1,增强与免疫治疗的协同效应。
**肿瘤坏死因子α(TNF-α)**类细胞因子也被引入PDC设计。NGR-hTNF由靶向新生血管的NGR肽与TNF-α融合而成,其中NGR结合肿瘤血管上过表达的CD13,TNF-α则改变内皮屏障功能、降低间质压,从而促进化疗药物在肿瘤中的渗透。在多项临床试验中,NGR-hTNF与多柔比星、卡培他滨、奥沙利铂等药物联合使用,显示出可控毒性与良好抗肿瘤效果,适用于结直肠癌、卵巢癌、小细胞肺癌等。然而,其III期随机研究未在总生存(OS)上显示显著优势,但在部分亚组患者中观察到PFS与OS改善。
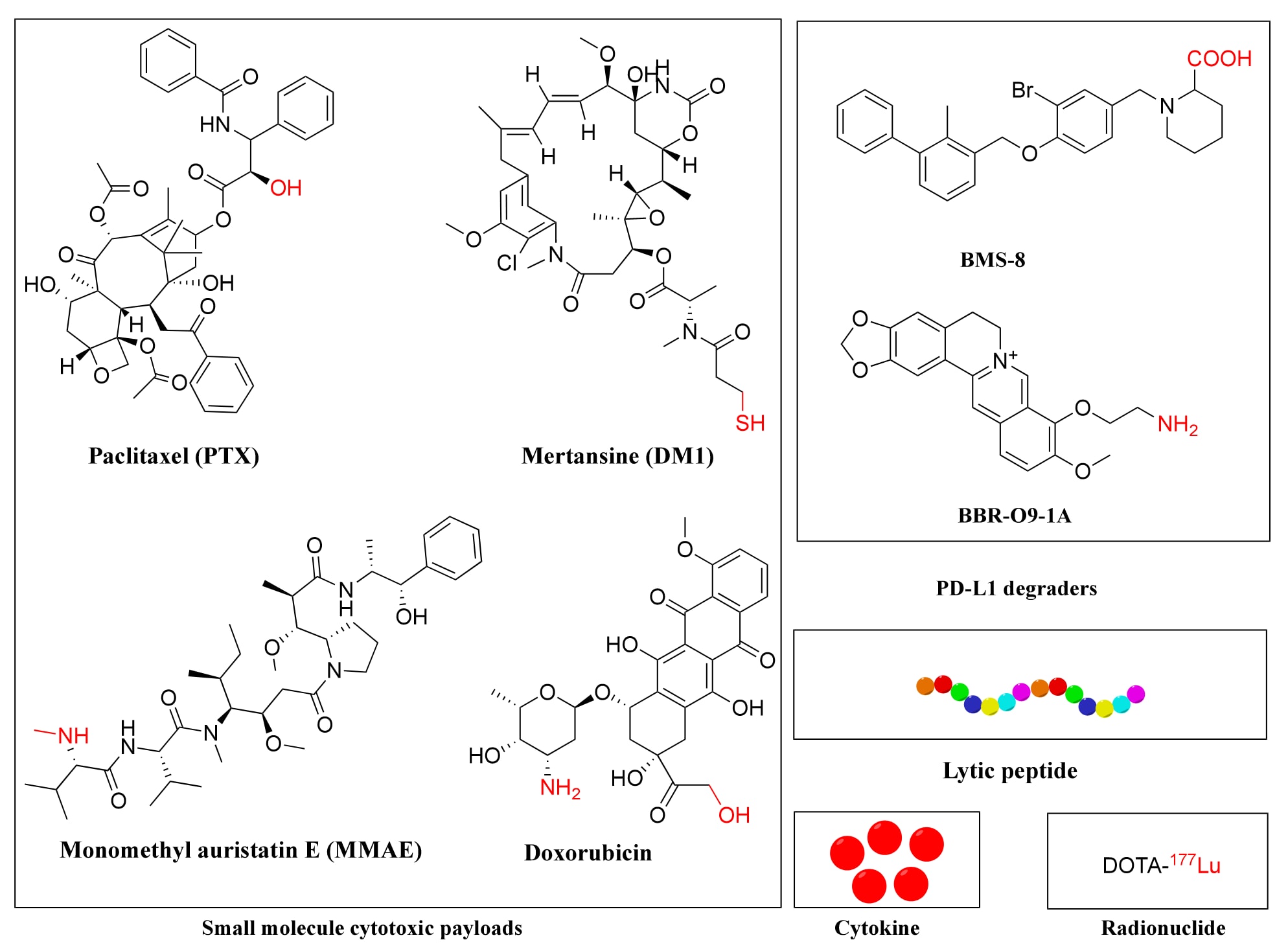
图9 | PDC代表性载荷的化学结构。 载荷与连接子上的功能基团以红色标注。
4 PDC的临床应用
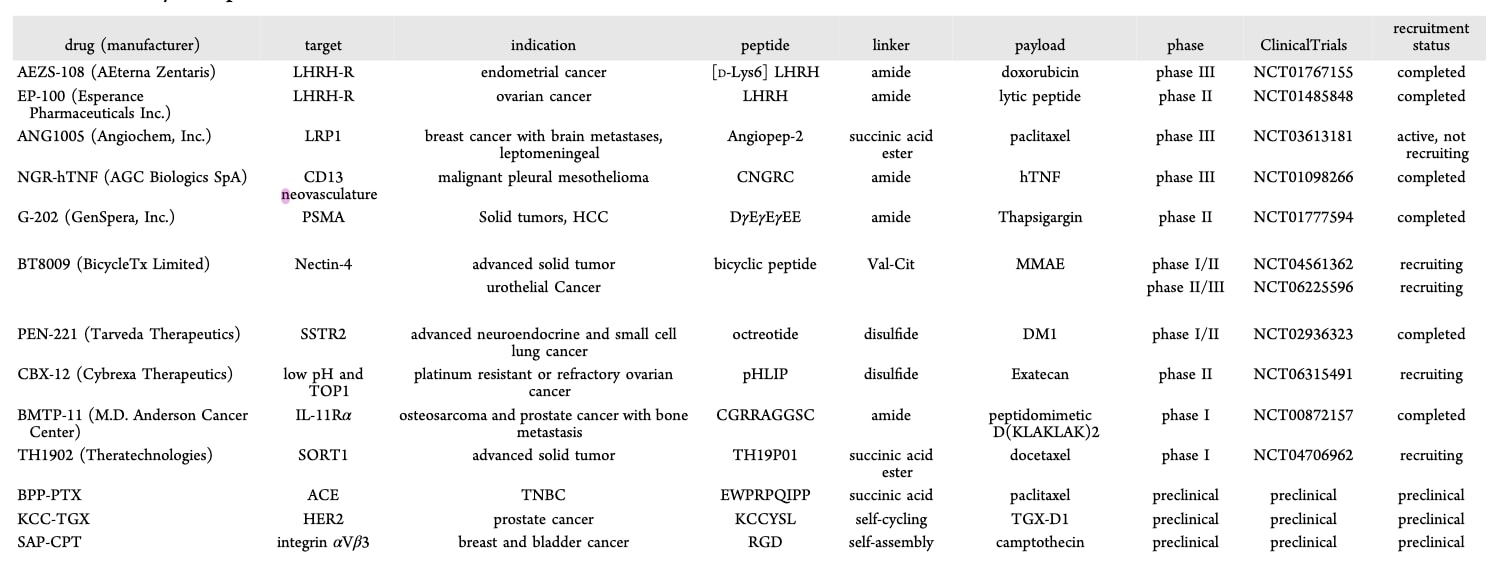
除已上市的Lutathera与Pluvicto外,多种PDC药物正在积极进行临床前或临床试验研究(Table 1)。
促黄体生成素释放激素(LHRH, 又称GnRH)是一种由下丘脑分泌的激素肽,其受体在卵巢癌、子宫内膜癌(EC)、前列腺癌及乳腺癌中均高表达。AEZS-108(zoptarelin doxorubicin,也称AN-152或ZEN-008)由阿霉素与LHRH激动剂化学偶联而成,特异性靶向LHRH受体。其酰胺连接子可被溶酶体内的酰胺酶选择性切割。由于卵巢与子宫内膜为激素依赖性器官,而晚期或复发性EC在铂类治疗后的标准疗法尚不明确,因此这些受体为AEZS-108提供了特异性靶点。一项多中心II期临床试验表明,AEZS-108在晚期或复发性LHRH受体阳性EC患者中展现出良好的抗癌活性与低毒性。另一项II期研究也显示其在去势及紫杉类耐药的前列腺癌中具有可接受的安全性与疗效。然而,III期随机对照试验(NCT01767155)显示,AEZS-108在局部晚期、复发或转移性EC的二线治疗中,并未优于阿霉素或dostarlimab在OS与PFS上的表现。近期研究认为,这一失败可能源于连接子不稳定与载荷效力不足。改进后,新型PDC D-Cys6-LHRH vedotin在卵巢癌中的生物活性与选择性显著优于AEZS-108(GI50分别为4 nM vs 453 nM)。
G-202 (mipsagargin)由靶向PSMA的肽段(DγEγEγEE)与小分子药物thapsigargin偶联而成。在一项II期多中心单臂研究中,G-202作为索拉非尼治疗失败后晚期HCC的二线药物,表现出抗肿瘤活性并延缓疾病进展。
研究还发现,sortilin (SORT1)在大部分乳腺癌、卵巢癌与子宫内膜癌中高表达。另一方面,生长抑素是一种能抑制多种激素分泌并调控消化与细胞生长的肽,其受体SSTR2在多种实体瘤中高水平表达。为克服天然生长抑素的局限性,研究者合成了多种类似物,如Lanreotide、Vapreotide与Octreotide。基于此,PEN-221被设计为一种PDC,由微管靶向剂DM1偶联至Octreotide的C端侧链,特异性作用于SSTR2。其二硫键连接子在肿瘤细胞内的还原环境中被切割,释放自由药物。前临床模型与SSTR2阳性小细胞肺癌患者的研究均显示出显著抗肿瘤活性。
TH1902 (Sudocetaxel Zendusortide)是一种新开发的SORT1靶向型多肽-多西他赛偶联物。在SORT1阳性的卵巢癌与三阴性乳腺癌异种移植小鼠模型中,其抗癌活性显著优于游离多西他赛。机制研究发现,TH1902能够激活cGAS/STING通路,增强抗PD-L1免疫介导的肿瘤杀伤作用。这是首次证明TH1902的抗肿瘤作用部分依赖于免疫肿瘤微环境的调控。
表1 | 临床试验中代表性PDC的汇总。
5 结论与展望
PDC因其独特性质而展现出巨大的药物研发潜力。尽管已取得显著进展,但其临床广泛应用仍受限制,主要原因在于细胞靶向能力不足及易受酶降解。通过多价性、双特异性或卤代修饰等策略,可以显著增强其靶向性;通过环肽、双环肽、白蛋白结合、D-氨基酸或非天然氨基酸替换、N-甲基化、拟肽(peptidomimetics)与拟肽类(peptoids)的策略,则能够提升PDC的稳定性与循环半衰期。然而,这些方法各有局限,目前尚无单一策略能实现理想PDC所需的全部特性。例如,D-氨基酸替换虽可改善抗蛋白酶稳定性,却可能降低生物活性。
近期的研究证明,高效CPP的开发有助于突破生物学屏障并提升PDC疗效,而将CTPs与CPPs、SAPs与SRPs结合,也能显著改善PDC的生物活性。与此同时,新型联合治疗方案不断涌现。例如,负载适配体-药物偶联物(ApDC)的细菌在胰腺癌治疗中表现出显著优势,细菌凭借穿透能力与ApDC的靶向与毒性作用实现协同,且能延长ApDC的血清稳定性至48小时,提升肿瘤部位药物浓度。
在肿瘤特异性特征的利用上,KRAS突变癌因缺乏传统结合位点长期被视为“不可成药”靶点。但研究发现,白蛋白结合药物可利用KRAS突变癌的巨胞饮特性,实现泛KRAS肿瘤靶向。新型靶向巨胞饮的PDC已被开发,在KRAS突变癌中表现出增强的细胞凋亡与更高的载荷累积。另一方面,含非天然氨基酸的共价肽能靶向非半胱氨酸残基(如组氨酸),并结合平坦蛋白表面等“不可成药”靶点,将进一步拓展PDC在癌症治疗中的应用。
总之,PDC领域正迅速发展。肽段设计、连接子化学与新型载荷在提升疗效中发挥核心作用。随着对细胞生物学理解的深入,PDC有望在未来成为药物研发的重要方向,并在多种疾病的治疗中展现更大价值。