Nat. Chem. Biol. 2025 | 利用计算技术推动抗体–药物偶联物的开发
今天介绍的是发表在 Nat. Chem. Biol. 上的一篇 Perspective,主题是利用计算技术推动抗体–药物偶联物(ADCs)的开发。ADCs 通过将小分子化疗药物的高毒性与单克隆抗体的高特异性结合,成为癌症治疗中极具前景的策略。然而,ADC 的研发过程依然复杂而艰巨,尤其是如何在保持抗体稳定性的同时,合理设计连接子、偶联位点和药物/抗体比例(DAR),以实现高效且安全的药物递送。
文章指出,传统实验手段往往难以捕捉抗体与药物负载的柔性和动态特性,而计算方法能够在早期阶段发挥关键作用。其中,基于物理的分子动力学模拟可以揭示抗体–抗原相互作用、糖基修饰及结构稳定性等原子层面细节;而机器学习方法则凭借丰富的单抗与ADC 数据库,实现快速预测并辅助制剂优化。作者还展示了 SILCS 等预计算模拟如何高效筛选潜在偶联位点和连接子设计,从而显著减少实验迭代。
总体来看,该研究强调了三维结构信息与计算模型结合的重要性,展望了物理模拟与机器学习在 ADC 研发中的协同潜力,为未来更高效的个体化抗体药物设计提供了新方向。

Croitoru, A.; Orr, A. A.; MacKerell, A. D. Harnessing Computational Technologies to Facilitate Antibody–Drug Conjugate Development. Nature Chemical Biology 2025, 21 (8), 1138–1147. https://doi.org/10.1038/s41589-025-01950-z.
0 摘要
抗体–药物偶联物(ADCs)是一类在癌症治疗中极具潜力的疗法。其核心思想是将已知化学药物的毒性与单克隆抗体的特异性结合,从而在提升疗效的同时最大程度地减少不良副作用。尽管已有多种ADC成功进入市场,但其研发过程仍然面临重大挑战,其中一个关键原因是缺乏能够充分反映单克隆抗体及药物负载物固有柔性的三维结构信息。
作者探讨了包括机器学习与基于物理的计算方法在内的多种计算技术如何帮助解析实验数据,并在药物连接子类型、偶联位点、药物/抗体比例等关键问题上提供预测,从而减少ADC研发中的设计迭代次数。同时,还举例说明了如何利用基于物理的三维分子建模与模拟来加深对模型ADC的理解,并推动更高效的ADC设计。
1 引言
细胞毒性疗法长期以来一直是癌症治疗的标准手段,但其副作用范围广泛。为了克服这一问题,能够靶向恶性细胞的治疗方式应运而生,它们通过精准递送毒性分子来最大化疗效并降低副作用,是个体化医疗中的重要工具。在这类疗法的研发中,单克隆抗体尤其具有吸引力,因为它们对特定靶标蛋白和细胞具有高亲和力与特异性,且易于开发。将单克隆抗体与细胞毒性分子结合,就形成了抗体–药物偶联物(ADC)。
ADC的概念最早在1958年的研究中被提出,随后经过多年的探索,1983年首次开展了人类研究,结果显示抗体能够将有效负载物递送至作用位点,为单克隆抗体作为递送工具的治疗应用打开了大门。到2024年,已有13种ADC获批临床使用,更多候选正在进行临床试验。然而,ADC的研发依旧面临挑战,不仅要考虑单克隆抗体本身的问题,还要应对药物负载通过共价连接带来的特定复杂性。
为了加速研发,三维结构信息结合计算方法展现出巨大潜力。作者将讨论能够促进ADC研发的计算方法,并介绍基于物理模拟的一些案例。抗体研发中通常需要关注稳定性、黏度和聚集性等因素,虽然临床上已有大量单抗成功应对了这些问题,但每一种新抗体都会带来独特挑战。计算方法可在早期做出预测,从而减少实验的时间和成本。一旦确定了作为靶向载体的单抗和细胞毒性药物负载,就需要明确药物与蛋白的结合位点和连接子设计,并重新评估负载药物对制剂性质的影响。迄今为止,专门针对ADC研发的计算研究仍然有限。该综述将概述相关计算方法,包括连接子的优化、偶联位点选择和药物/抗体比(DAR)的确定,并通过模型ADC展示基于物理模拟所能提供的信息,最后展望未来计算方法在ADC研发中的改进方向。
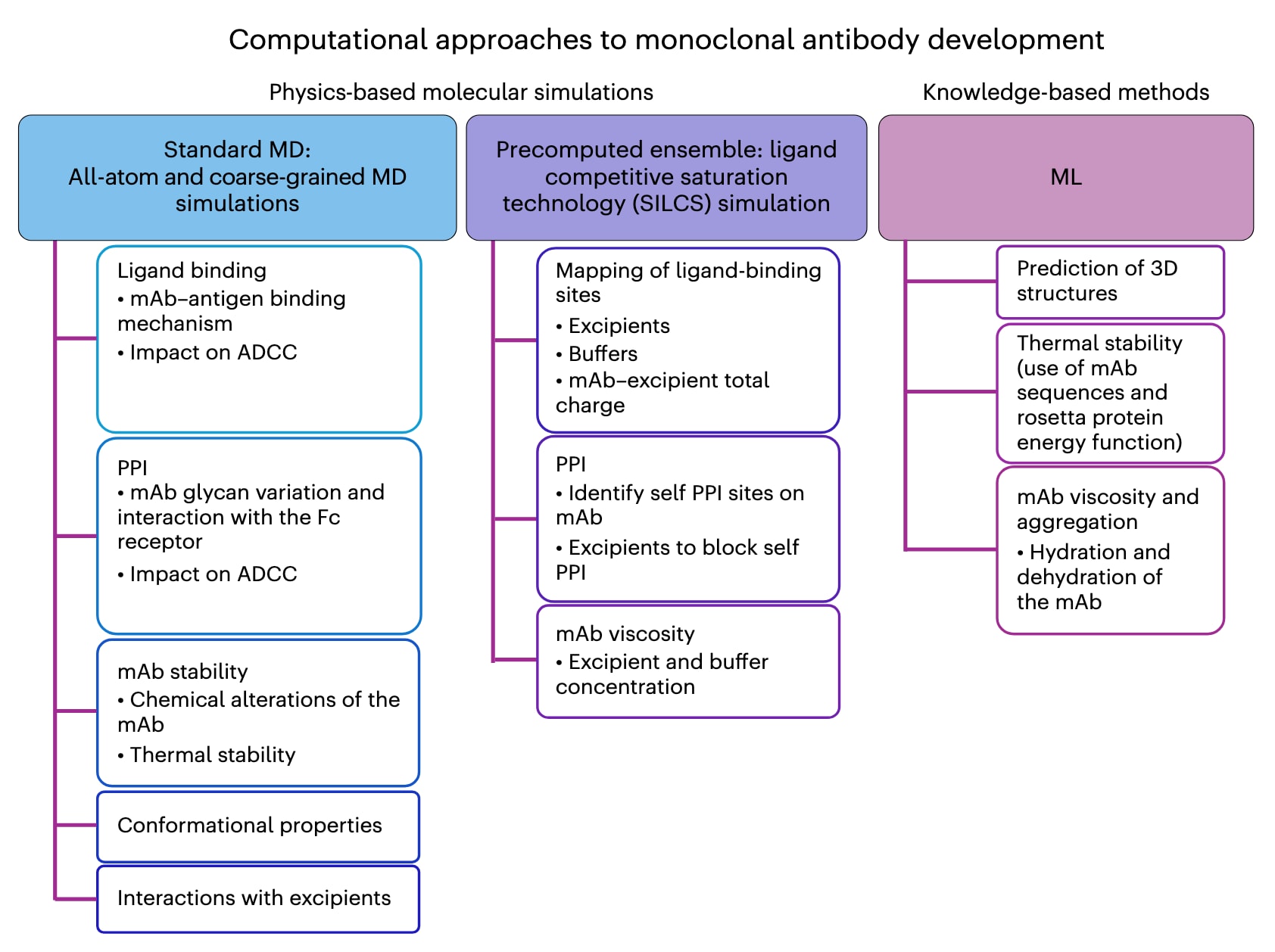
图1 | 通过计算方法获得的单克隆抗体性质示意总结。 基于物理的模拟方法利用描述物理原理的数学模型,通常基于经典牛顿力学和经验力场。这类方法可分为两类:① 标准分子动力学(MD)方法,需要对每一个新的化学实体进行显式模拟以进行预测;② 预计算的集合方法,如 SILCS,其通过初始MD模拟生成数据,后续对新化学实体的预测则依赖该数据,而无需额外的MD模拟。基于物理的模型可应用于任何新的ADC,而无需先验性质知识。基于知识的方法则通过现有数据训练数学模型,随后对新体系进行预测,但预测能力受限于新化学实体与训练数据的差异。这类方法包括快速发展的机器学习(ML)方法。mAb,单克隆抗体。
2 单克隆抗体研发中的计算方法
用于推动单克隆抗体研发的计算方法主要包括基于物理的和基于知识的两大类(图1)。前者以分子对接和分子动力学(MD)模拟为主;后者则从传统的定量构效关系(QSAR)方法延伸到机器学习(ML)。全原子MD模拟能够提供单抗的分子层面图景,包括其构象特性以及与溶质(如赋形剂、缓冲液和离子)的相互作用。此外,粗粒化MD方法可用于研究更长时间尺度和更大体系规模的问题,例如单抗的自聚集现象。
近年来,基于ML的方法逐渐应用于单抗的开发和制剂研究,尽管在ADC上的具体应用仍相对有限。ML工具包括突破性的蛋白质三维结构预测方法(如AlphaFold和RoseTTAFold),以及专门针对单抗开发的算法。最新的AlphaFold 3甚至能够预测蛋白质上的小分子结合位点。这些进展以及其他ML方法,得益于单抗领域丰富的实验数据资源,包括ADC数据库等,从而推动了ML方法的快速发展。然而,由于训练此类模型需要多样体系的大量新实验数据,未来ML方法的快速扩展可能会受到资源需求的限制。
2.1 基于物理的方法
在单克隆抗体研究中,基于物理的方法主要以全原子分子动力学(MD)模拟为核心。相关应用包括:考察诱导契合与锁钥模型在抗体–抗原相互作用中的作用机制;探索抗原结合Fab片段后如何通过变构效应调控Fc区,从而改变其与Fc受体的结合并影响抗体依赖的细胞毒性(ADCC)。结合实验的MD研究已揭示了Fc–Fcγ受体相互作用的原子细节,并指出糖基化变异如何改变Fcγ受体结合及ADCC效应。对Fc–糖链–受体相互作用的分子层面理解,有助于通过糖工程手段调控ADCC与补体依赖性细胞毒性。
2.2 体稳定性的物理与知识驱动方法
MD模拟也被用于研究单抗的化学修饰,例如天冬氨酸消旋和天冬酰胺脱酰胺化等过程,它们会影响蛋白的降解稳定性。关于单抗及VHH单域抗体热稳定性的MD研究,则揭示了天然接触与VH–VL结构相对取向的重要作用。与此同时,机器学习(ML)方法也被用于预测单抗的热稳定性,输入特征包括抗体序列和Rosetta能量函数的参数。与MD相比,ML方法具有显著的计算效率优势,但其适用性受到训练数据覆盖范围的限制;而MD则能推广到任意序列,尽管其准确性依赖于势能函数的合理性。此外,还必须验证MD模拟是否已充分收敛,以确保结果的可靠性。
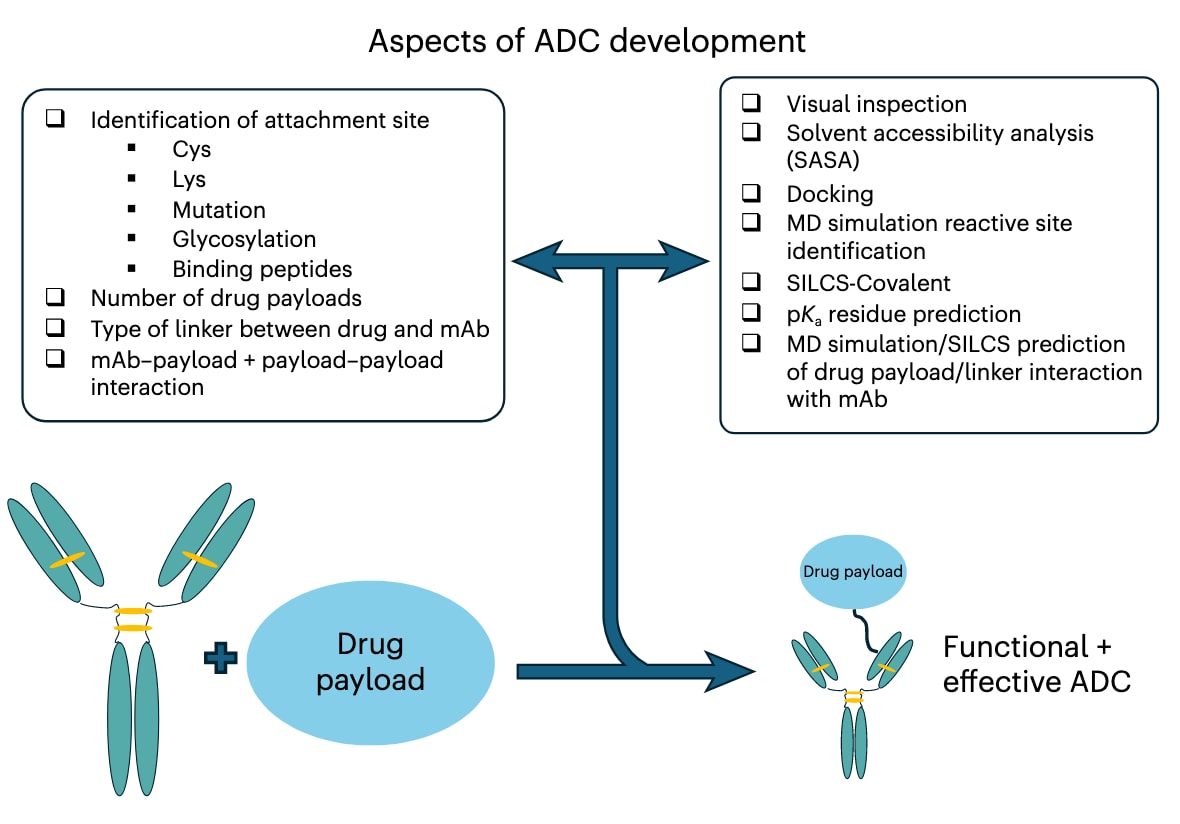
图2 | ADC开发的各个方面示意图。 左侧列出ADC开发时需考虑的设计因素,包括单克隆抗体本身的性质及药物负载。右侧列出在决策过程中可能使用的计算方法与信息类型,这些方法有助于实现功能性和有效的ADC。计算方法可通过改进实验数据的解释和提供预测,减少实验次数,从而节省时间与经费。
2.3 黏度与聚集的建模
早期的分子动力学(MD)研究主要用于探讨单抗的胶体特性,例如优先相互作用系数(Γ23)。近期,MD与机器学习(ML)结合用于预测Γ23值。ML方法还被用于预测单抗的高黏度潜力,并设计突变以降低黏度,同时也被应用于单抗的黏度与聚集性预测。总体而言,ML方法速度更快,但受到训练数据的限制,难以提供分子层面的细节,例如赋形剂和缓冲液如何影响单抗的聚集和黏度。而这类信息对于优化制剂至关重要,却是MD方法能够直接获得的。MD的另一优势在于其能够捕捉到部分展开的中间态,而这些中间态往往会促进聚集。
2.4 配体竞争饱和(SILCS)方法
由于每一种新ADC变体(包括制剂变化)都需计算大量构象,MD模拟往往计算开销巨大。为此,提出了配体竞争饱和(SILCS)方法。该方法结合大正则蒙特卡洛与MD模拟,预先生成单抗及其周围溶质和水分子的构象集合,并据此计算功能基团的自由能亲和性模式(即网格自由能 FragMaps)。这些 FragMaps 可被反复使用,以快速定量评估小分子与单抗的相互作用,例如映射赋形剂和缓冲液的结合位点及亲和性。对于ADC而言,这种方法能够预测毒性“弹头”、连接子及偶联位点对单抗–药物相互作用的影响。此外,该方法还能计算蛋白–蛋白自相互作用(PPI),为高黏度与聚集性提供机制解释。
结合PPI热点区域,SILCS对赋形剂和缓冲液结合模式的映射能够指导制剂设计,从而加速单抗进入临床的进程。扩展方法 SILCS-Biologics 已被用于解释赋形剂对治疗性单抗Fab黏度变化的影响,并能区分电荷相近的赋形剂结合位点,例如赖氨酸与精氨酸,其中赖氨酸倾向结合于PPI相关位点,从而解释了实验观察到的黏度下降。SILCS还被应用于NISTmAb Fab的分析,结果显示,预测的赋形剂结合位点数量和分布与PPI热点区域的关系可用于预测单抗的黏度与稳定性。
更近期的研究表明,SILCS映射结果与NMR数据一致,已被用于HIV疫苗单抗的制剂开发,以及血清白蛋白的赋形剂结合分析。这些成功部分归因于SILCS能够考虑局部构象异质性,这对于合理处理互补决定区(CDRs)的需求尤其重要,因为CDRs很可能主导单抗制剂的设计要求。此外,SILCS-Biologics还能预测有效蛋白电荷,这是单抗开发中的关键因素。蛋白电荷越高,蛋白–蛋白排斥作用越强,从而限制PPI。该方法还能综合考虑制剂中物种的存在与浓度对总电荷的贡献,为不同制剂方案下的快速评估提供支持。
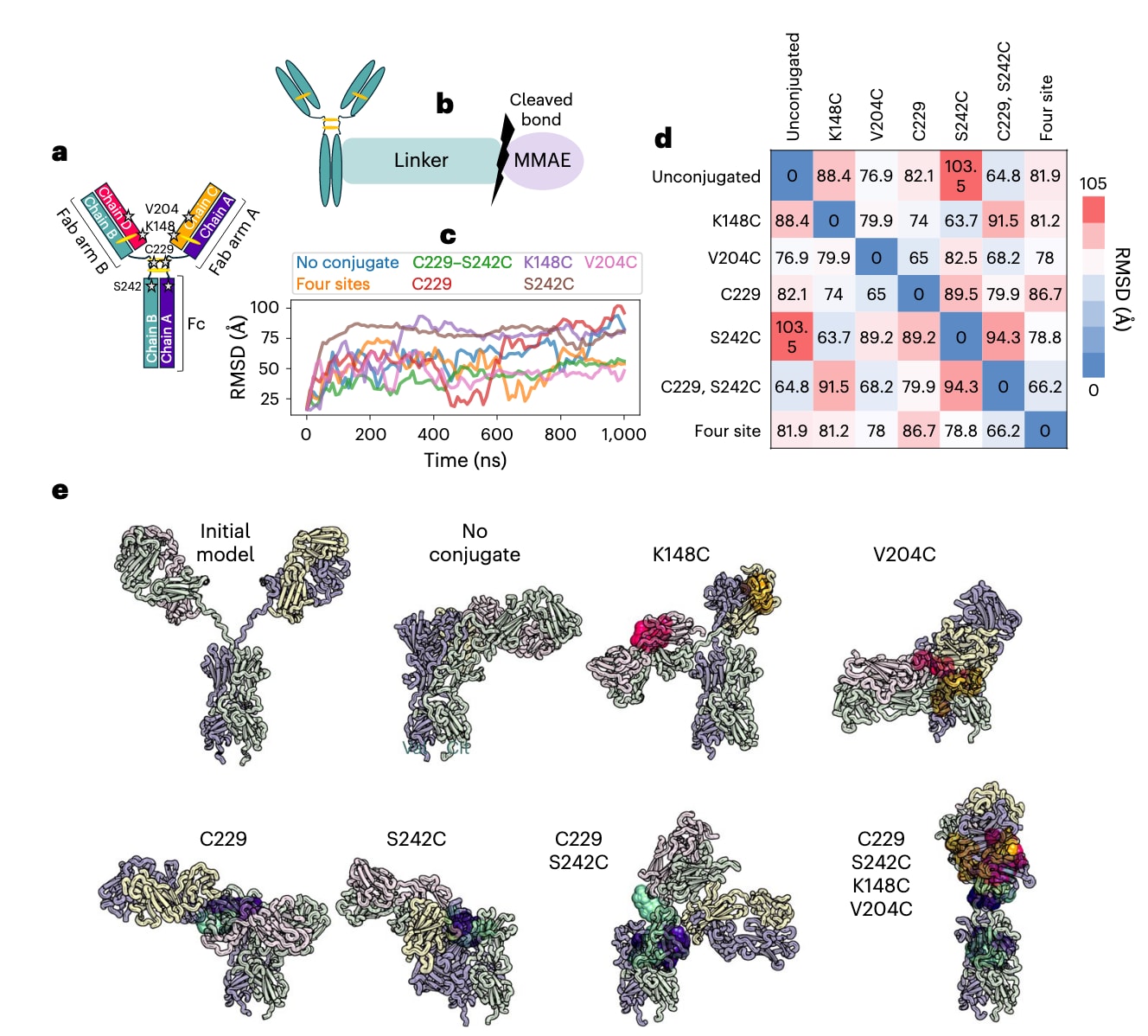
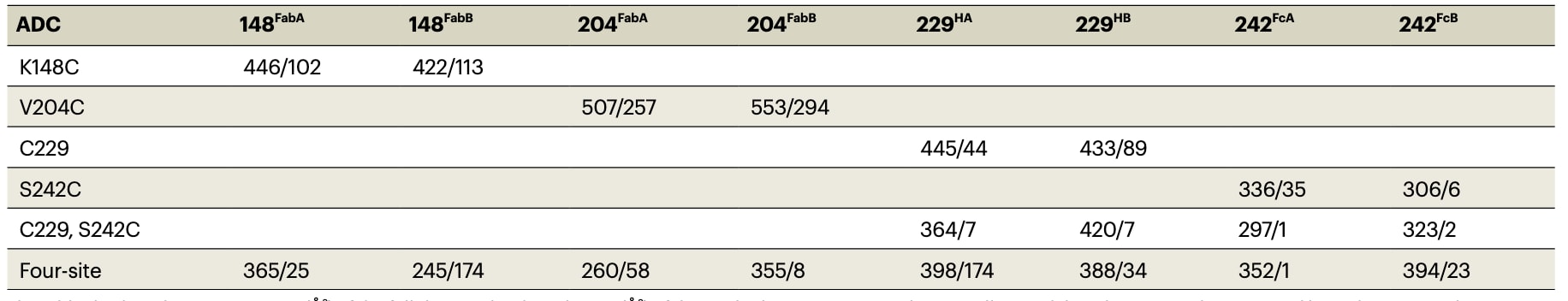
图3 | 单克隆抗体可在多个位点与药物负载偶联,从而影响ADC的动力学与结构特性。 a,单克隆抗体的示意图,显示偶联位点和二硫键,多肽链用不同颜色区分。b,完整药物负载的示意图,包括与单抗的共价连接、目标肽键及MMAE有效负载(化学结构见补充信息)。 c,MD模拟中Fab片段相对于Fc的RMSD(基于Fc非氢原子的对齐)。 d,7个体系Fab片段相对于Fc的二维RMSD矩阵(同样基于Fc非氢原子对齐,取自MD模拟的最终快照)。 e,完整单抗的初始模型、非偶联单抗及ADC的MD最终结构。ADC的对齐基于初始模型中所有Cα原子。颜色方案与a中的多肽链一致,共价连接的药物负载(彩色原子球)与对应链同色。补充图1展示了基于所有单抗Cα原子对齐的版本。
3 ADC 开发的独特方面
抗体–药物偶联物(ADC)的开发,除了要选择合适的单克隆抗体和毒性“弹头”外,还涉及**连接子类型、偶联位点的选择以及药物负载数(DAR)**的确定。这些因素不仅影响药物与抗体本身的相互作用,还可能导致相邻药物之间的相互作用,从而带来潜在的生物学影响(图2)。关于弹头类型与修饰策略,已有综合性综述进行过系统总结,此处不再展开。
3.1 偶联位点的选择
ADC 开发中的一个难点是如何在最大化疗效的同时,保持抗体稳定性。常用的偶联位点是半胱氨酸和赖氨酸,因为它们具有较高的反应活性,并且有成熟的化学修饰试剂可用。以 IgG1 为例,其仅包含有限数量的半胱氨酸残基,例如铰链区的两个二硫键以及轻链与重链之间的二硫键。这一特征虽然便于控制偶联位点和DAR,但直接修饰铰链区的半胱氨酸往往会引发稳定性问题。为克服这一点,研究者开发了“双功能重桥接”方法,通过在二硫键上进行再连接,实现了曲妥珠单抗(trastuzumab)DAR=4的稳定构型。
相比之下,赖氨酸在抗体中数量更多,提供了更大的修饰灵活性,但其特异性和DAR控制较差。为此,可以通过突变引入新的残基以实现位点特异性修饰,或者通过肽段结合引导反应性连接子的方法实现选择性偶联。此外,基于糖链的偶联策略也已被提出,例如通过 Fab 区糖基进行修饰,或者借助糖工程在抗体序列中引入新的糖基化位点。这类方法还能改变ADC与Fc受体的相互作用,进而影响抗体依赖的细胞毒性(ADCC)。
3.2 计算方法的助力
尽管上述实验策略已取得进展,但往往依赖反复的尝试与优化。计算方法在此可大大减少试错成本。静态结构的可视化与溶剂可及性分析(如利用 Chimera 软件)能够快速识别潜在偶联位点,再结合分子对接方法预测连接子–药物的结合位点。例如对接实验曾预测某Fab区域能稳定结合“vc-seco-DUBA”连接子,随后通过定点突变将该区域残基改为半胱氨酸,实验成功获得多个ADC,从而验证了预测的准确性。
进一步地,MD模拟的动力学特征可以揭示静态结构中不可见的“隐藏”反应位点,例如 SILCS-Covalent 方法便可识别埋藏的半胱氨酸残基。此外,残基的 pKa 值能够反映其亲核性,MD 或 ML 方法均能预测残基 pKa,从而为偶联位点筛选提供指导。
一个有趣的基于物理的研究提出利用非共价加合物进行ADC开发。通过对接与MD模拟,研究者确定了小分子 4-巯基乙基吡啶在抗体上的结合位点,该分子可作为锚点连接两种不同的“连接子–药物”结构(MAGNET-Gem 和 MAGNET-Fluor)。实验结果显示,这两种负载在体外能够稳定结合抗体,DAR约为6,并在体内展现出优于裸抗体的抗肿瘤活性。
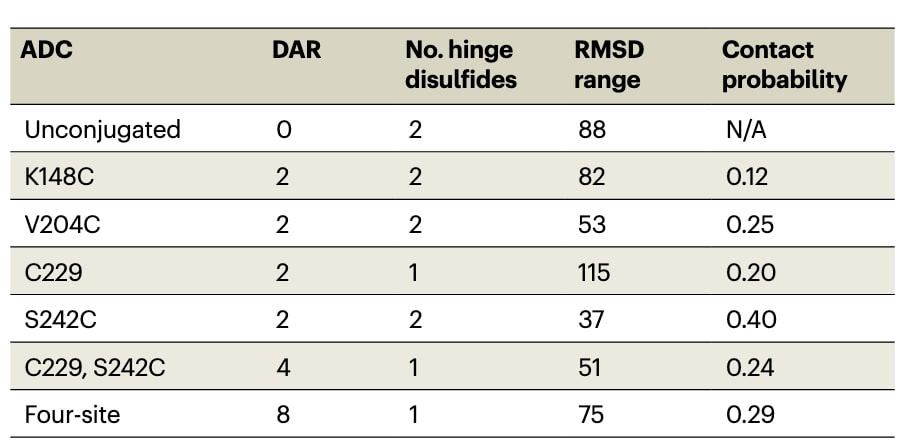
表1 | 基于NISTmAb和MMAE的模型ADC(来源于ADC brentuximab vedotin)。
3.3 连接子与药物“弹头”对负载相互作用的影响
ADC 开发中的一个关键问题在于药物负载本身的性质,它直接决定了其与单克隆抗体以及相邻负载分子的相互作用方式。若负载与抗体之间存在过强的相互作用,可能会限制药物的可及性。实验表明,更具疏水性的药物以及更高的 DAR(药物/抗体比值)往往导致抗原结合能力下降,而DAR升高虽能提升细胞毒性,却会削弱结合特异性。此外,有研究显示,降低药物的疏水性可改善整体疗效。
基于物理的计算方法能够预测连接子类型与偶联位点如何影响“负载–抗体”及“负载–负载”之间的相互作用,从而间接决定负载的可及性。这类信息对于优化药物释放、避免药物负载与Fab–抗原结合的竞争具有重要价值。
表2 | 不同ADC的完整偶联体溶剂可及表面积(SASA)和木瓜蛋白酶B靶点的部分可及表面积(PASA)。
4 三维结构建模以促进 ADC 的早期设计
在 ADC 的开发过程中,需要对大量设计方案进行预测,从而缩小进入实验验证阶段的候选范围。除了根据生物学靶点选择单克隆抗体和药物“弹头”外,设计空间还由**连接子类型、偶联位点以及药物/抗体比值(DAR)**共同决定。这些变量可以直接映射到单克隆抗体的三维结构上,并借助分子动力学(MD)模拟,或结合 SILCS 方法进行分析,以评估其对生物学反应的潜在影响。通过计算筛选出的最优组合往往是那些能最大限度减少负载与抗体本身及相邻负载之间不利相互作用,同时又能保证连接子充分暴露、利于药物释放的方案,这类组合才值得进入后续实验环节。
4.1 ADC 开发的三维结构需求
在 ADC 研究中,三维结构信息是应用物理建模方法的基础。尽管目前尚未有完整 ADC 的实验结构报道,但单克隆抗体的三维结构往往可以通过X射线晶体学、冷冻电镜或人工智能预测方法(如 AlphaFold 和 RoseTTAFold)获得。然而,仅有一个静态结构不足以驱动 ADC 设计,因为抗体及药物负载都具有高度柔性。因此,必须考虑构象集合,这一问题正是基于物理的 MD 模拟所擅长解决的。
4.2 基于物理的 ADC 分子模拟
一个展示 MD 在 ADC 设计中效用的模型体系是 NISTmAb。其 Fab 与 Fc 片段已有实验晶体结构报道,因此可以通过对齐这些已知结构来构建完整抗体模型(见补充说明2)。在某些情况下,Fab 与 Fc 也可以分别进行模拟,再将结果组合分析,SILCS 方法即采用这种思路。
在 ADC 建模中,首先需要将药物负载通过化学键连接到抗体的特定位点(补充说明3)。通过这种方式,可以生成并评估大量实验上可行的连接子类型、偶联位点和 DAR 组合(图3a)。例如,NISTmAb 可通过可切割连接子与 MMAE 偶联,这种设计正是 brentuximab vedotin 中的实现方式,其连接子包含与半胱氨酸共价偶联的马来酰亚胺片段,以及可被蛋白酶切割的缬氨酸–瓜氨酸–对氨基苯甲酸氨基甲酸酯片段(图3b)。利用计算建模,可以生成多种不同 DAR 的构型,如 DAR=4(C229–S242C)或 DAR=8(四位点偶联)(表1与图3a)。
4.3 MD 模拟与分析
物理模拟依赖于势能函数(力场)来描述分子运动。常用的力场包括 Amber、OPLS-AA 和 CHARMM36(用于大分子),以及 CGenFF、OPLS 或 GAFF2(用于药物负载)。相应的软件工具有 CHARMM、Amber、NAMD、GROMACS 和 OpenMM。模拟必须在水溶液环境中进行,通常显式加入水和离子(如100 mM KCl)。
在正式生产模拟之前,需要经过充分的平衡阶段,以使 ADC 从初始模型松弛至合理构象。平衡期可能需要约 100 ns,在此期间可观察到 Fab 相对于 Fc 的初始松弛(图3c),构象变化幅度可达 100 Å(通过 RMSD 衡量)。这些变化反映了整体抗体由初始“Y”型构象松弛为其他 Fab–Fc 相互作用程度不同的构象(图3e)。完成平衡后,才进入生产模拟阶段(例如 101–1000 ns),用于分析 ADC 的各种结构与动力学性质。
4.4 构象多样性与药物负载的影响
在生产阶段的 MD 模拟中,ADC 持续发生构象变化,包括 Fab 相对于 Fc 的运动(图3c,表1,RMSD 范围)。模型体系采样到不同的构象,这一点可通过最终结构的 RMSD 比较(图3d)以及直观观察(图3e,补充图1)得到验证。显然,ADC 能够呈现一系列不同的构象,而这些构象会受到负载药物的影响,并可能带来 生物学层面的后果。例如,在 S242C ADC 中,负载药物与 Fab A 发生相互作用,导致 RMSD 范围较小;但在 C229–S242C 与四位点 ADC 中,由于额外负载的存在,S242C 连接的药物位于 Fc 中央“甜甜圈孔”处,从而失去与 Fab A 的相互作用(图3e,补充图1),使体系的柔性增加(表1)。整体而言,大多数 ADC 的 RMSD 范围与游离单抗相当或更低,说明负载药物在一定程度上限制了抗体的构象柔性。C229 ADC 是个例外,可能与失去一个铰链区二硫键有关。
4.5 药物–药物相互作用
负载间的相互作用在多个负载共存的体系中尤为显著。以三类在 S242C 位点连接药物的 ADC 为例:其 RMSD 范围从 S242C ADC 的 37 Å,增至 C229–S242C ADC 的 51 Å,再到四位点 ADC 的 75 Å(表1)。进一步分析显示,S242C ADC 中 MMAE 与抗体的接触概率最高(表1)。直观观察发现,S242C ADC 中负载药物彼此之间以及与 Fab A 都存在相互作用;但在 C229–S242C 或四位点 ADC 中,新增负载扰动了 S242C 药物的作用,导致其接触概率下降。这些差异表明,偶联位点与 DAR 的选择可用于调控抗体的构象特性。
4.6 药物可及性与胞内释放
药物负载需要通过 组织蛋白酶B(cathepsin B)切割才能释放,因此了解连接子的可及性对 ADC 设计至关重要。理想情况下,切割位点应尽可能暴露,以便高效水解;若追求受控释放,则中等程度的可及性更为合适。常用指标包括全构象的溶剂可及表面积(SASA),以及切割位点(如缬氨酸、瓜氨酸及相邻氮原子)的蛋白可及表面积(PASA)(图3b)。不同 ADC 的 SASA 和 PASA 变化明显(表2)。整体趋势是,单位点负载(DAR=2)的可及性通常高于 DAR>2 的情况,但也存在例外。例如,C229 负载在单点 ADC 中可及性较大,但在 C229–S242C ADC 中显著减小,而在四位点 ADC 中又显著增加,甚至超过单点情况。相反,S242C 负载在三类 ADC 中均表现为低可及性,提示该位点的偶联可能不利于 cathepsin B 介导的药物释放。此类差异为选择合适的 连接子–位点–DAR 组合提供了重要信息。
4.7 SILCS 预计算抗体构象集在 ADC 设计中的作用
由于对每个 ADC 变体进行完整 MD 模拟的计算代价过高,SILCS-Biologics 提供了一种高效替代方案。其核心是基于预计算的 自由能网格(GFE FragMaps),可快速评估不同连接子类型与偶联位点下药物的结合模式与亲和力。以 MMAE 为例,研究者分别在 Fab 与 Fc 的 FragMaps 上进行全面对接(图4a,b),预测所有可能的结合位点。与 MD 模拟得到的接触残基(图4c,d)比较后发现,两者吻合良好,说明 SILCS 能有效预测药物结合位点与可连接药物数目,而无需为每种组合执行长时间 MD。此外,SILCS 还能识别 MD 中未采样到的潜在结合位点(图4a,b),从而避免在接近天然结合区域的残基上进行偶联。
SILCS 还可以进一步预测药物或连接子是否会影响抗体与抗原的相互作用,尤其是在互补决定区(CDR)区域。此外,FragMaps 所提供的信息还能用于 辅料选择,从而辅助 ADC 制剂开发。通过减少实验迭代次数、提升数据解读效率,SILCS 为早期 ADC 设计提供了有力支持。
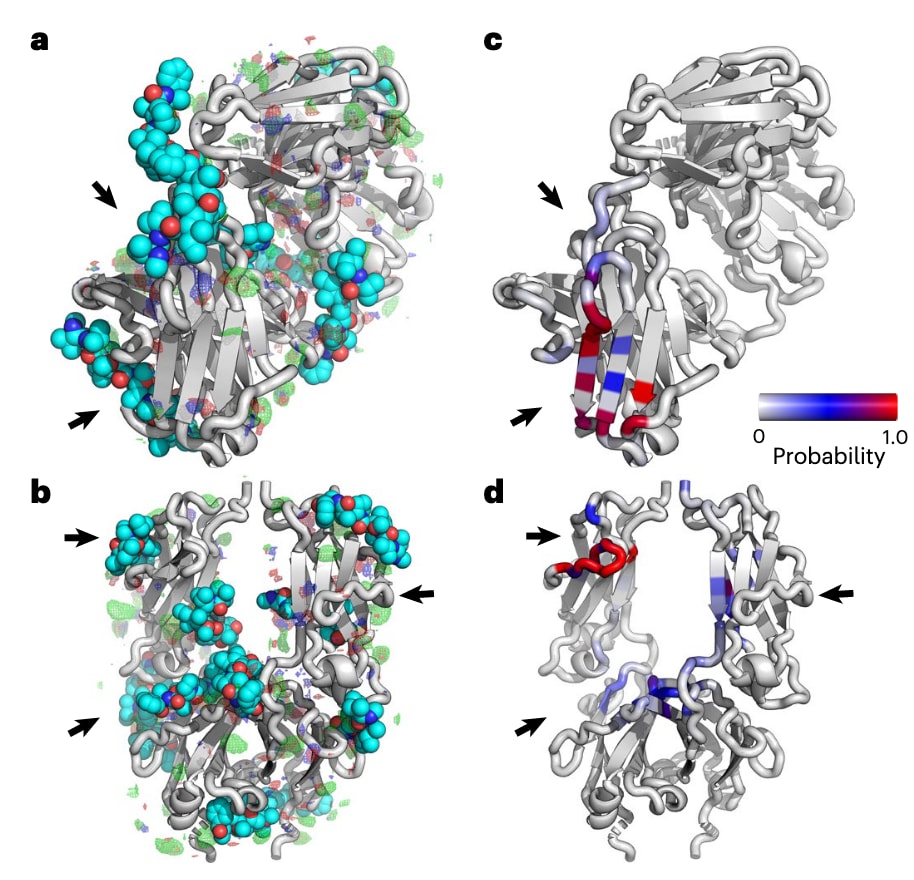
图4 | SILCS识别所有可能的有效负载结合位点,避免进行多次耗时的MD模拟。 a,c,Fab A:展示了SILCS FragMaps以及MMAE有效负载的对接构象,预测结合自由能(LGFE)< −10 kcal·mol⁻¹(a),并显示了表1中所有模型ADC的MD模拟中与MMAE有效负载接触的残基(c)。b,d,Fc:展示了SILCS FragMaps及MMAE有效负载的对接构象(b),并显示了表1中所有模型ADC的MD模拟中与MMAE有效负载接触的残基(d)。接触残基基于5 Å非氢原子距离阈值。箭头指示SILCS识别的MMAE结合位点(a,b),与MD模拟识别的接触区域一致(c,d)。SILCS GFE FragMaps以网格显示:非极性(绿色,−0.9 kcal·mol⁻¹)、氢键供体(蓝色,−0.6 kcal·mol⁻¹)、氢键受体(红色,−0.6 kcal·mol⁻¹),能量值为可视化的阈值。
5 结论与展望
基于物理的计算技术使得 三维结构信息 能够在 ADC 早期开发中得到应用。正如前文所示,这类方法能够以预测的方式应对设计挑战,包括连接子类型、偶联位点和 DAR 等关键因素。通过减少药物与单抗本体或药物之间的直接相互作用,从而改善连接子区域的暴露度并促进药物释放,研究者可以从大量候选中筛选出最优的连接子–位点–DAR 组合进入实验验证。此外,这些方法还能帮助解释首代 ADC 的实验数据,加速迭代优化。随着开发逐步推进到体内实验甚至临床阶段,当出现意外问题时,研发人员也能快速回溯并重新评估此前已进行过计算的组合,探索新的实验路径。
基于物理的方法的一大优势是,它们可以用于此前从未研究过的体系。从 Fab、Fc 或全抗体的三维结构出发,MD 模拟和相关方法能够为全新体系提供可观测数据,为 ADC 开发“起步”。不过,单抗的分子体积给 MD 带来挑战,且势能函数的准确性也限制了模拟结果的可靠性。好在 GPU 的普及与计算效率提升,使得大规模 MD 模拟愈发可行;加之并行化、独立轨迹采样与增强采样技术的发展,进一步拓展了时间尺度与统计能力。另一条有效途径是 SILCS 的预计算集,它能在实验前快速评估大量组合,从而降低实验负担。
在精度方面,势能函数若能显式处理 电子极化效应,将特别有利于带电体系的建模,例如药物负载、缓冲液或辅料。随着方法精度与适用性的提升,基于物理的模拟在 ADC 早期开发中的作用还将持续扩大。
与此同时,机器学习方法 因能够实现快速预测而备受关注,理论上可涵盖 ADC 开发的各个方面。但它们受限于训练数据的覆盖范围。虽然已有单抗与 ADC 数据库可供使用,但直接支持药效、稳定性或制剂相关建模的数据仍然有限。如果能获得来自药企的更全面数据,将极大促进相关模型的开发。
未来,物理模拟与机器学习结合的潜力尤为值得期待。基于物理的模拟可生成图结构特征,如偶联位点的空间分布、负载可及性、特定 warhead 的结合位点及其亲和力等,并以此构建图神经网络等深度学习模型。若能配合充足的实验训练数据,这类模型有望实现强大的预测能力,从而推动 ADC 开发从早期制剂扩展到更广泛的应用阶段。
总之,预计 物理模拟与机器学习方法在 ADC 开发与制剂中的应用将显著扩展,并在未来产生更深远的影响。