STTT 2024 | G蛋白偶联受体(GPCRs):结构、机制与药物发现的最新进展
**今天介绍的是发表在 Signal Transduction and Targeted Therapy(STTT)2024 年的一篇综述。**GPCR 作为人体中数量最庞大、最具药理价值的膜蛋白家族,长期占据 FDA 批准药物靶点的“第一梯队”。随着冷冻电镜、XFEL、NMR 与分子动力学等技术的快速演进,人类在过去十年间获得了前所未有的大量 GPCR 原子级结构,从正构激动剂到变构调节剂,从G蛋白偏向性到细胞内变构口袋,结构生物学不断刷新我们对这一超家族的认知。
该综述系统梳理了 GPCR 结构解析的最新突破、激活与信号调控机制、受体多态性、突变与疾病关联,并进一步聚焦小分子药物发现,深入解析了近年 FDA 批准的代表性正构药物与最新报道的变构调节剂复合结构。文章不仅总结 11 类可成药变构位点,还提出“机制驱动药物设计”的理念,为未来开发高选择性、高效能且具偏向性的 GPCR 治疗分子提供了清晰路线图。

Zhang, M.; Chen, T.; Lu, X.; Lan, X.; Chen, Z.; Lu, S. G Protein-Coupled Receptors (GPCRs): Advances in Structures, Mechanisms and Drug Discovery. Signal Transduction and Targeted Therapy 2024, 9 (1), 88. https://doi.org/10.1038/s41392-024-01803-6.
0 摘要
G蛋白偶联受体(GPCRs)是人体中数量最多的膜蛋白家族,也是重要的药物靶点类别,在维持多种生理过程方面发挥关键作用。激动剂或拮抗剂、正构或变构效应、偏向性信号或平衡信号等多种特点共同构成了GPCR动态性质的复杂性。该研究首先回顾GPCR在结构解析、活化机制与功能多样性方面的最新进展,随后聚焦于GPCR药物发现,通过解析近五年内获得美国食品药品监督管理局批准的正构类药物的精细药物−靶点相互作用及其潜在机制,阐明其设计逻辑与作用基础。特别地,文章对已解析的与合成小分子变构调节剂复合的GPCR结构进行了最新分析,用以揭示关键的受体−配体相互作用与变构调控机制。最后,研究强调了广泛存在的GPCR可成药变构位点如何推动基于结构或基于机制的药物设计,并提出未来通过设计双位点配体提升该受体家族治疗潜力的发展方向。
1 引言
G蛋白偶联受体(GPCRs)是细胞表面膜受体中最大的一类,上千个基因编码了这一超家族。它们共享由七段跨膜螺旋(7TM)以及三条胞内环与三条胞外环连接而成的保守结构。GPCR属于构象高度动态的蛋白,能够响应多种胞外信号,包括光子、离子、脂质、神经递质、激素、肽类与气味分子,以此介导关键的信号转导功能。由于胞外配体结合位点与胞内信号事件之间相隔约40Å,GPCR的信号传递本质上属于变构过程。
随着蛋白工程、X射线晶体学、冷冻电镜(cryo-EM)的发展,以及XFEL与NMR等技术的引入,人们对GPCR结构与动态特性的理解得到了革命性提升。这些研究揭示配体−受体相互作用、构象变化以及信号复合体的特征,大幅推动对受体活化、正构与变构调控、偏向性信号及二聚化等过程的深入解析。
GPCR被外源刺激激活后,主要通过异源三聚体G蛋白与arrestin传递信号,产生第二信使并触发下游通路,从而在细胞内形成多样化的信号谱系,这是GPCR功能多样性的基础,并在感知、神经传递、内分泌调节等生理过程中发挥关键作用。然而,GPCR的突变或缺失会扰乱其正常功能,可能改变其固有活性、影响膜表达或干扰翻译后调控。深入阐明刺激−GPCR−效应器的耦合机制,以及如何精准调控GPCR功能障碍,将为开发高效、高选择性或偏向性调控的分子提供重要启示。
**迄今为止,大约34%的FDA批准药物以GPCR为靶点,且处于临床试验与前临床阶段的调节剂数量仍在快速增长。**正构配体通过与内源性配体竞争结合,有效改变GPCR活性与信号过程,但由于正构位点在亚型间序列保守,选择性往往成为难题,副作用也因此难以避免。作为替代或补充手段,单独靶向变构位点或同时作用于正构与变构位点能够克服这些不足。变构调节剂通常具有更高的亚型选择性与更低副作用。随着受体−配体相互作用的结构细节不断清晰,结构基础药物设计(SBDD)从片段到先导化合物的优化路径变得更加明确。此外,对变构位点的认识也可用于设计同时占据正构与变构位点的双位点配体。**与单独的正构或变构配体相比,双位点配体通常具有更高亲和力与更强选择性。**同时,阐明GPCR变构机制也为开发偏向性配体提供可行策略,例如偏向G蛋白或β-arrestin通路的变构调节剂。通过特定通路产生作用的双位点调节剂能够进一步减少副作用。
**该综述首先总结GPCR在结构解析、活化机制与功能多样性方面的进展。**为了进一步探讨GPCR药物发现的最新发展,文章分析了近期FDA批准的正构类药物的结合模式与靶点机制。随后对变构调节剂进行系统讨论,重点聚焦近年解析的与合成小分子变构调节剂结合的GPCR结构。**分析排除了肽与抗体类分子,针对变构位点在胞外前庭、跨膜区域与胞内表面的分布进行了系统归类,总结其关键结合模式与变构机制。**该综述旨在为基于结构或机制的药物设计以及未来双位点配体的开发提供深入洞察,以推动GPCR靶点的治疗潜力。
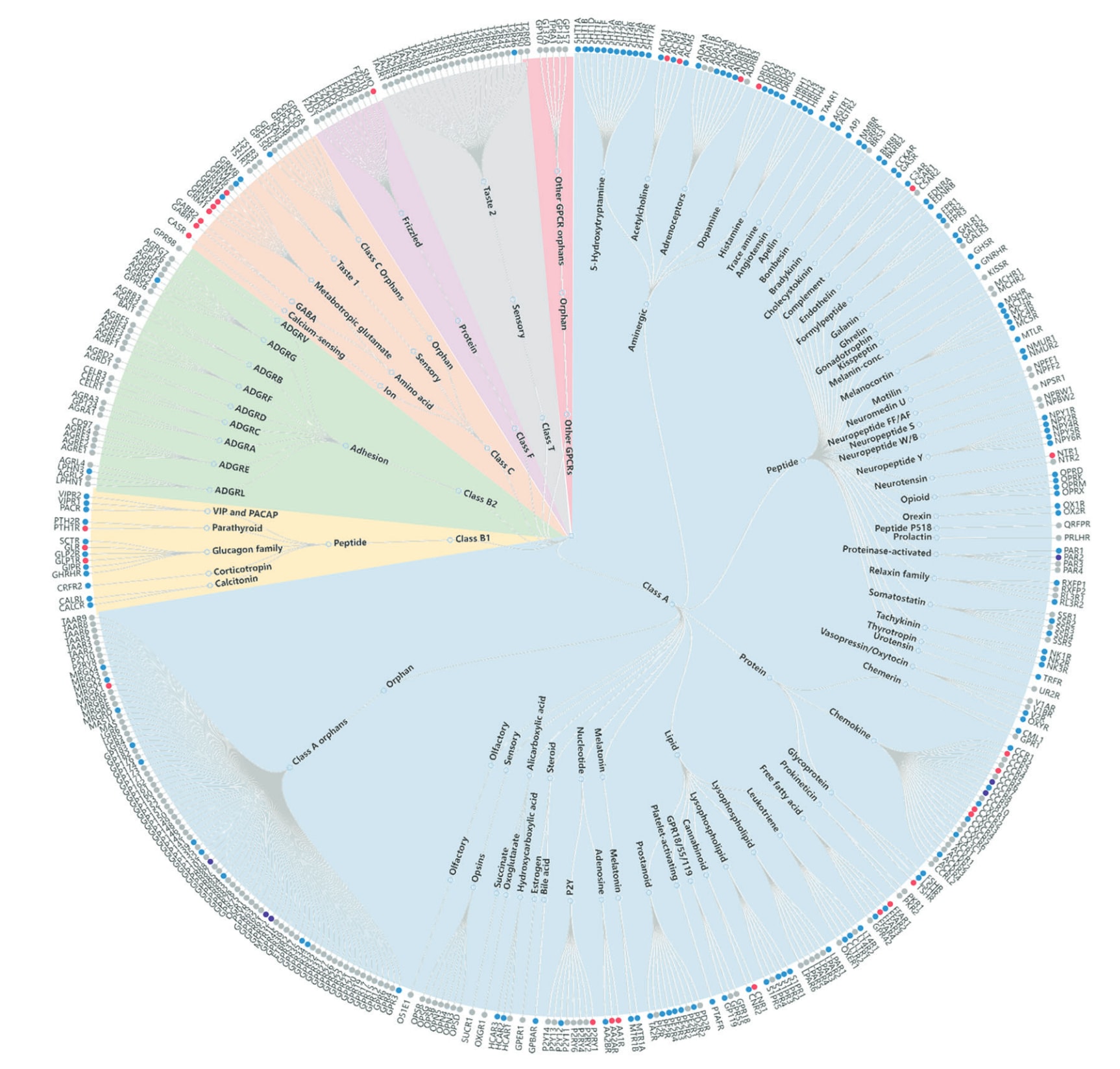
图1|GPCR系统进化树,展示已解析的与调节剂复合的GPCR结构。 节点以UniProt基因名称标注GPCR,并按照GPCR数据库的分类体系进行组织。与调节剂结合的GPCR结构以颜色标示。
2 GPCRs 结构进展
膜蛋白GPCR表达量低且构象高度灵活,这些特性曾长期阻碍其获得高分辨率衍射结构。视紫红质与β2肾上腺素受体(β2AR)的首次晶体结构分别于2000年与2007年解析。过去二十年中,蛋白工程与X射线晶体学技术不断进步,包括通过融合蛋白工程、抗体片段辅助结晶以及热稳定性突变等策略,成功解析了大量结合拮抗剂或激动剂的GPCR结构。然而,激动剂结合状态的GPCR往往停留在中间构象,因为完全活化构象通常需要G蛋白、G蛋白模拟物、构象特异性纳米抗体或mini-G蛋白等伴侣稳定。
首个GPCR−G蛋白复合物于2011年通过X射线衍射获得,但由于晶体学要求极高,GPCR−G蛋白复合物的结晶仍极具挑战。Cryo-EM的兴起为GPCR结构生物学带来突破。与依赖晶体的X射线方法不同,cryo-EM可直接观察以去垢剂或纳米盘溶解的GPCR,并能解析此前难以获得的完全活化状态与更大蛋白复合物,包括GPCR−G蛋白复合体。从此,结合胞内伴侣的GPCR的cryo-EM结构呈指数级增长。截至2023年11月,蛋白质数据库已收录554个相关复合结构,其中523个通过cryo-EM解析。
然而,无论晶体学还是cryo-EM,其捕获的通常是晶体化条件下最稳定、能量最低的构象,对于中间态与构象转换动力学的完整刻画仍存在空缺。晶体学、光谱学与模拟方法为GPCR构象动态提供互补信息。XFEL以其超高亮度与飞秒脉冲特性,能够避免辐射损伤,在飞秒尺度上解析近原子分辨率的结构,有望解决缺失的动态信息。NMR可在液相中探测GPCR的动态特征,其信号的数量、位置与形状对受体中稳定同位素“探针”微环境的变化高度敏感。DEER可测量两探针之间的距离分布,FRET作为“分子标尺”可探测标记之间的接近程度,从而推测构象状态及其相对丰度,但两者都只提供局部信息。分子动力学(MD)模拟则能在时间维度上追踪完整蛋白结构,捕捉构象中间态与转变路径。
GPCR结构生物学的整体进展揭示了配体−受体相互作用、构象变化与信号复合体的关键特征,为深入探索受体活化、正构/变构调控、偏向性信号传导与二聚化奠定了重要基础。
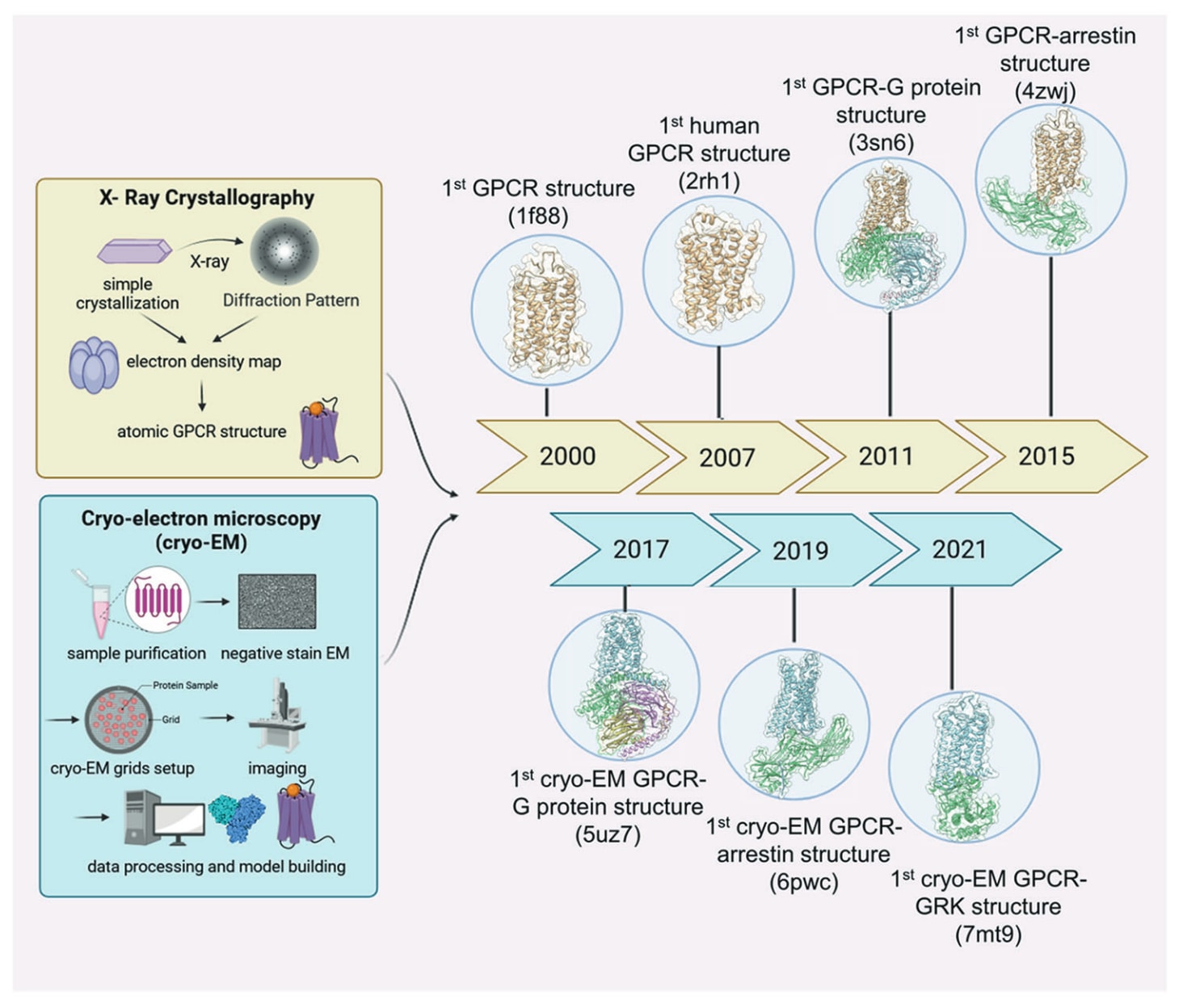
图2|使用X射线晶体学与冷冻电镜研究GPCR结构的重要进展时间线
3 GPCR的活化与信号转导机制
尽管GPCR本身的特性以及其激活刺激的来源可能差异巨大,但其下游信号主要依赖两类转导蛋白,即异源三聚体G蛋白与arrestin。人体G蛋白可分为Gs、Gi/o、Gq/11与G12/13四大类,超过一半的GPCR能够同时激活两种或以上的G蛋白,而不同G蛋白在效应与动力学上的差异使GPCR呈现高度多样化的耦联模式,从而在细胞内形成指纹式的信号特征,显著增加信号网络的复杂性。
**Gαβγ三聚体在结合GDP时保持不活化。**激动剂结合受体后,促使受体形成活化构象,开始招募并激活G蛋白。受体促进Gα亚基的GDP/GTP交换,使其与Gβγ分离,并因细胞内GTP浓度高而迅速结合GTP。Gα−GTP与Gβγ随后分别调控多类效应蛋白,Gα−GTP可激活或抑制酶类如腺苷酸环化酶、磷脂酶C或离子通道,而Gβγ也能够调控多条信号途径并与靶蛋白互作。效应蛋白被激活后会产生cAMP等第二信使。最终,Gα水解GTP形成GDP并重新与Gβγ结合,使G蛋白重新进入静息状态,完成一个循环。
为防止信号持续放大,活化的GPCR会被GRK在C端多位点磷酸化。这些磷酸化位点决定β-arrestin的结合能力,通过空间位阻使受体脱敏,并引发网格蛋白介导的内吞与泛素化。受体−arrestin复合体同时也是超过二十类激酶的支架,包括MAPK、ERK1/2、p38以及JNK等,从而引导G蛋白非依赖性信号通路。已知arrestin具有四种亚型,GRK也有多个亚型,其中arrestin1与arrestin4特异存在于视觉系统,而β-arrestin1与2可调控大量非视觉GPCR。
**GPCR最初被认为以单体存在,但随后发现其可形成同源或异源二聚体,在活化机制、药理特性与生物功能方面均有所不同。**近年的研究还显示GPCR可与多类单跨膜辅助蛋白结合,以调控其配体结合、转导蛋白耦联与信息传递功能。典型例子包括调控胰高血糖素受体的RAMP家族以及调控黑皮质素受体的MRAP家族,其与GCGR和MC2R的相互作用已被cryo-EM解析。
GPCR的活化特征包括TM6胞内端显著向外摆动,使受体内部腾出空间容纳下游转导蛋白。**多个保守结构基序参与活化过程,包括TM6的CWxP基序、TM7的NPxxY基序,以及TM3−TM6与TM3−TM7之间的离子锁。**此外,Na+作为A类GPCR的内源性负向变构调节剂,通过与TM1、TM2、TM3与TM7的氨基酸残基互作稳定不活化构象,而不同GPCR中其结合方式存在差异。
**配体通过稳定特定构象调控受体活性。**由于不同信号通路具备不同生理效应,能够选择性诱导有益通路的偏向性配体具有重要药理价值。**例如偏向G蛋白通路的μ阿片受体激动剂能够在增强镇痛的同时减少β-arrestin通路相关不良反应,这与吗啡形成鲜明对比。**代表性的偏向性配体包括TRV130、PZM21与SR-17018,它们已经进入临床使用或研究阶段。深入理解G蛋白、GRK与arrestin的耦联方式将为设计选择性调控特定信号路径的偏向性药物奠定基础。
在无激动剂条件下,部分GPCR仍具有不同程度的固有活性,而不同配体在激活或抑制同一受体方面的效能也差异较大。结合受体固有活性与配体效能,可将GPCR配体划分为完全激动剂、部分激动剂、拮抗剂与反向激动剂,而这些效能差别也会影响其治疗作用。
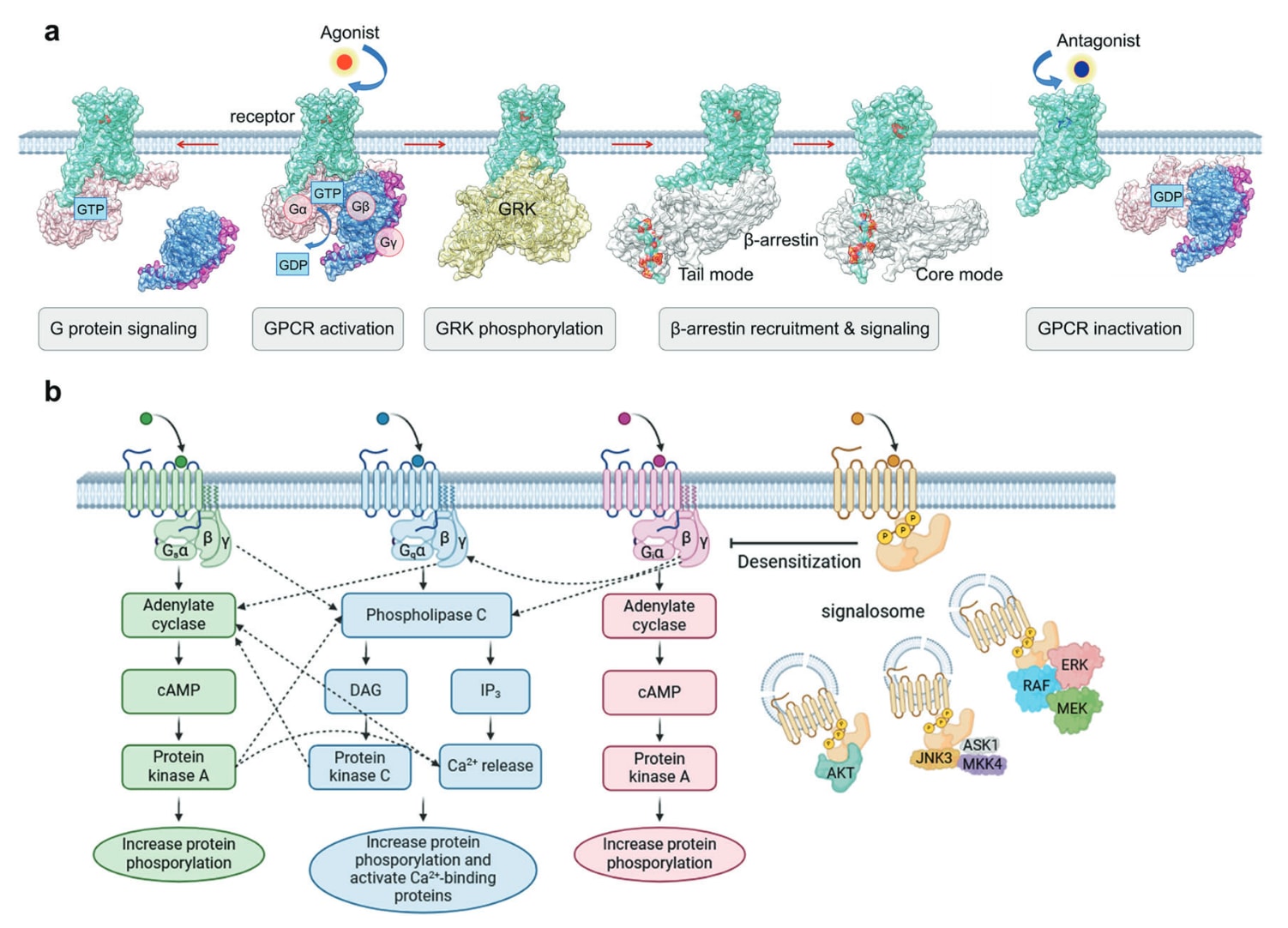
图3 a GPCR活化过程的示意图。激动剂(红色圆形)结合后,受体进入与G蛋白三聚体耦联的预激活状态,在此过程中G蛋白α亚基发生GDP/GTP交换,引发G蛋白解离并介导G蛋白信号通路。受体C端被GRK磷酸化后促进arrestin的募集与信号传导。当拮抗剂(蓝色圆形)结合时,受体被稳定在不活化状态。b Gs、Gq、Gi与arrestin下游信号通路的串联与交互。
4 GPCRs 功能多样性
**GPCR在结构与功能特征上可分为A、B、C、F与T五大类。**A类GPCR又称视紫红质样受体家族,是数量最多、研究最深入的一类。A类GPCR可依功能进一步划分为氨能、肽类、蛋白类、脂质类、褪黑素类、核苷酸类、类固醇类、二羧酸类、感觉类以及孤儿受体等多个亚群,其相关适应症涵盖高血压、心血管疾病、呼吸系统疾病、抑郁症以及精神疾病等。
B类GPCR分为分泌素样(B1)与黏附型(B2)两类。B1受体具有体积较大的胞外结构域,B2受体则具有独特的长N端结构域与可诱导自身蛋白切割的结构域。GLP-1R与GCGR是近年来最受关注的B1类受体,在调控血糖稳态与脂质代谢中具有核心作用;B2类受体则参与调控感觉、内分泌以及胃肠系统。
C类GPCR即谷氨酸受体家族,特点包括巨大的胞外结构域、保守的VFT结构、富含半胱氨酸的受体结合域以及依赖二聚化实现活化的机制。以代谢型谷氨酸受体(mGluRs)为代表的C类GPCR在临床转化中走在前列,其相关生理功能与癌症、偏头痛、精神分裂症及运动障碍等密切相关。
F类GPCR由10个frizzled受体(FZD)与一个SMO受体组成,具备保守的CRD区域,参与Hedgehog与Wnt信号通路,因此多与肿瘤、纤维化以及胚胎发育相关。目前药物开发主要集中于SMO,而FZD仍具有广阔的探索空间。
值得注意的是,调控味觉感知的TAS2R受体虽在结构上与A类GPCR相似,但其序列同源性低于20%,因而被归入全新的T类GPCR,这也进一步扩展了对GPCR家族整体的认识。
4.1 GPCR在感觉、神经传递与内分泌调控中的作用
4.1.1 Rhodopsin、TAARs与TASRs在感觉感知中的作用
GPCR最显著的生理功能之一,是介导光觉、味觉、嗅觉与信息素感知等多种感觉信息。视紫红质是脊椎动物视觉激活的第一步,也是A类GPCR的典型代表。在吸收光子后,其正构配体视黄醛会在皮秒尺度内发生构象翻转,从而迅速触发信号传播至G蛋白、cGMP磷酸二酯酶或cGMP门控离子通道。视黄醛与受体之间的共价键结合,以及其瞬时构象变化与信号放大过程,为理解GPCR在感觉系统中的高效性提供了范例。
嗅觉受体可分为气味受体(ORs)与微量胺相关受体(TAARs),是研究嗅觉信息编码的重要载体。最新研究揭示TAARs识别胺类气味分子的通用机制,并给出嗅觉受体“组合编码”的结构基础,同时明确了mTAAR9对Gs与Golf的选择性耦联,为解析哺乳动物嗅觉识别机制提供关键突破。除G蛋白外,嗅觉受体的下游信号还涉及腺苷酸环化酶与cAMP门控离子通道,使其成为仍具潜力的研究方向。
味觉受体(TASRs)是调控味觉这一核心生理感知的重要GPCR体系。从生理与药理学角度,味觉受体研究十分活跃。I型味觉GPCR通过形成异源二聚体介导甜味(TAS1R2/TAS1R3)与鲜味(TAS1R1/TAS1R3),而II型受体TAS2R为单体,主要调控苦味。当味物质结合受体后会激活下游第二信使,引发细胞去极化并敏化瞬时受体电位(TRP)通道,最终将信号传入大脑味觉皮层。由于传统GPCR表达技术难以适用于TAS2R,其结构解析长期受限,未来在结构层面的突破将显著推动其生理学研究。
4.1.2 μOR与CBR在神经传递中的作用
当前神经系统药物的主要需求集中在神经性疼痛缓解、抑郁与精神疾病治疗以及帕金森病的干预等方面。μ阿片受体(μOR)已有超过半个世纪的研究历史,其在外周神经系统与中枢神经系统中的镇痛机制已被深入揭示。例如,在伤害感受神经元中,μOR通过与TRPV1、H1R与NK1R等受体互作,能够减少痛觉相关物质的释放并降低Ca2+的产生;而在脊髓背角神经元中,μOR可调控5-HT受体、甘氨酸受体以及去甲肾上腺素受体,激活下行疼痛抑制通路。为了在发挥镇痛作用的同时减少呼吸抑制与成瘾等副作用,研究者已先后开发正构偏向调节剂、变构调节剂以及双位点调节剂。
大麻素受体(CBR)同样是参与神经传递与神经性疼痛病理的重要靶点。CB1R主要分布于中枢神经系统神经元的突触前末端,其激活可抑制神经递质释放与痛觉信号传递;CB2R则在免疫细胞中高度表达,其激活能够抑制促进痛觉敏化的炎症因子。非选择性正构激活CB1R与CB2R的化合物在多种动物模型中可产生明确的镇痛效果与睡眠改善作用,而选择性正向变构调节剂如ZCZ011等则因避免类似大麻类药物的不良反应而成为更具前景的配体类型。
4.1.3 GLP-1R与GPR120在内分泌调控中的作用
内分泌疾病已成为21世纪最重要的健康问题之一。多类与代谢密切相关的GPCR通常由能量代谢物或底物激活,是感知内分泌失衡的重要分子传感器。其中,GLP-1R与GPR120(亦称游离脂肪酸受体4)是治疗2型糖尿病与肥胖的关键靶点。
GLP-1R的内源性配体GLP-1能够在胰腺α细胞中减少胰高血糖素的分泌,并在β细胞中促进胰岛素的释放。相较之下,GPR120在结合ω3多不饱和脂肪酸后可减少脂肪组织炎症并对抗胰岛素抵抗,其与Gq/11耦联后会激活PI3K/Akt通路,从而促进脂肪细胞对葡萄糖的摄取。
随着GLP-1R激动剂利拉鲁肽成为FDA批准用于2型糖尿病与肥胖治疗的代表药物,更多与内分泌调节相关的GPCR靶点如GPR35、GPR40、GPR41、GPR43、GPR81与GPR119有望进入药物研发视野。
4.2 受体多重耦联特性与不同信号通路之间的交叉调控
GPCR将胞外刺激转化为胞内信号,从而调控细胞功能与表型。这些胞内信号通路彼此交织,可增强或削弱相关反应,这种现象被称为“交叉调控”。由此形成的GPCR信号网络呈现高度多重耦联,导致调控范围更广、选择性更低,并可能产生副作用。多重耦联与交叉调控可发生在三个层面,即受体本身、G蛋白与β-arrestin,以及下游效应器。
受体层面的多重耦联主要源于异源二聚体的形成,二聚体既可以由同一受体家族的不同亚型组成,也可以跨家族形成。典型例子是GABAb(1)与GABAb(2)的异源二聚体,两者以单体形式存在时均无功能,而二聚体能够调控GIRK钾通道。另一成熟案例是腺苷受体与多巴胺受体之间的广泛互作,A1A与A2A受体的激活会降低D1与D2受体的多巴胺结合能力。可同时结合腺苷受体与多巴胺受体的双价配体进一步证明了异源二聚化的存在与功能意义。这些二聚体被认为共享同一G蛋白池,从而重分配其对G蛋白的耦联方式并重塑信号格局。基于此,通过两个GPCR之间的直接交叉调控,有可能设计作用于一个受体的配体来调节另一靶点的亲和力与效应,尽管其药理特征仍有待明确。
第二层级涉及GPCR对Gs、Gi、Gq、G12以及β-arrestin1与2的招募。在这一阶段,从高度选择性的耦联到显著的多重耦联均可观察到。MD模拟研究显示,通过改变GPCR胞内界面的构象,工程化突变能够改变受体对非典型G蛋白的耦联,说明GPCR胞质结合口袋的“动态结构可塑性”是G蛋白多重耦联的基础。因此,通过突变、正构或变构调节剂,施加精细且远程的影响以调控胞内结合界面,成为实现G蛋白选择性的主要策略。
第三层级即下游效应器的多重耦联,其与G蛋白交叉调控高度相关。通常情况下,Gs、Gi与Gq分别激活腺苷酸环化酶、抑制腺苷酸环化酶与激活磷脂酶C。然而,当多类G蛋白在膜附近同时被招募时,所释放的βγ亚基可在不同信号通路之间“互换”,从而增强其他通路的反应。随后第二信使彼此之间发生磷酸化、激活或抑制,形成精细调控网络(图3b)。尽管相关研究已非常丰富,但如何精准控制GPCR的多重耦联仍未完全明确。
4.2.1 GPCR突变对人类疾病的影响及其治疗学意义
GPCR不仅广泛参与多种生理过程,其基因突变也与多类人类疾病密切相关,这凸显了开展GPCR基因组学研究的重要性,并具有直接的治疗学意义。目前已鉴定出超过2350个GPCR基因突变,它们是60余种人类单基因遗传病的主要致病因素(图4a)。其中,错义突变占比最高,超过60%;小片段插入或缺失位居第二,占比超过15%。
4.2.2 GPCR功能异常中突变作用的分类
GPCR突变对受体功能的影响可大致分为功能获得型(GoF)与功能缺失型(LoF),分别对应生理功能的过度增强与降低。近年的研究进一步揭示这两类突变背后的多样化机制。相比野生型受体的正常活化模式,活化型与失活型突变在药理学上的影响主要体现在三个方面。首先,突变能够改变受体内部的微开关级联反应,诱导受体偏向活化或不活化构象,从而影响其固有活性与下游效应器的募集。其次,突变会以直接或间接方式影响受体的表达水平,从而改变其在细胞内的运输、降解与再循环过程。再次,部分突变会影响配体的效能、特异性或识别特征,通过调控受体构象分布、改变下游耦联方式或影响受体二聚化来发挥调节作用。
需要强调的是,并非所有变异都具有致病性,这一事实为突变的另一种分类方式提供依据,即将其区分为“驱动型”与“乘客型”(图4b)。近年来,基于大量临床突变数据及其对应的GPCR功能异常,计算方法正逐渐兴起,用于预测突变在GPCR相关疾病中的驱动能力,为理解疾病机制与开发治疗策略提供新的方向。
4.2.3 GPCR突变与人类疾病的关联
由GPCR突变引起的基因改变是多种单基因遗传病的重要驱动因素。其中较为明确的案例包括SMO受体的错义突变导致基底细胞癌,MC4R的错义与无义突变导致肥胖,以及FSHR的错义突变引发卵巢过度刺激综合征。多数突变位点具有高度保守性,因此在进化过程中处于相对优势地位。由此可见,在评估GPCR突变与人类疾病之间的病理关联时,将某一残基的进化保守性纳入考量会使预测更加有效。
4.2.4 GPCR相关病理的治疗意义与干预策略
GPCR相关病理的治疗策略主要包括对症治疗与病因治疗。许多GPCR功能异常最终会导致具有靶器官抵抗特征的内分泌疾病或肿瘤,因此可通过激素补充或化疗药物来缓解病理表型。然而,更前沿的治疗理念正逐渐转向病因干预。GPCR中的错义突变往往导致蛋白折叠与翻译后修饰的错误,从而引发运输障碍,此时可采用药理伴侣作为潜在治疗方案。对于由无义突变或移码突变导致的受体截短,可通过RNA干扰、基因替代以及CRISPR/Cas9等基因编辑手段恢复受体的完整性,只要突变受体至少保留前三个跨膜螺旋。设计多肽或小分子调节剂则是恢复受体药理功能的最直接方式,但在多突变背景下开发成本高昂,仍是难以解决的问题。
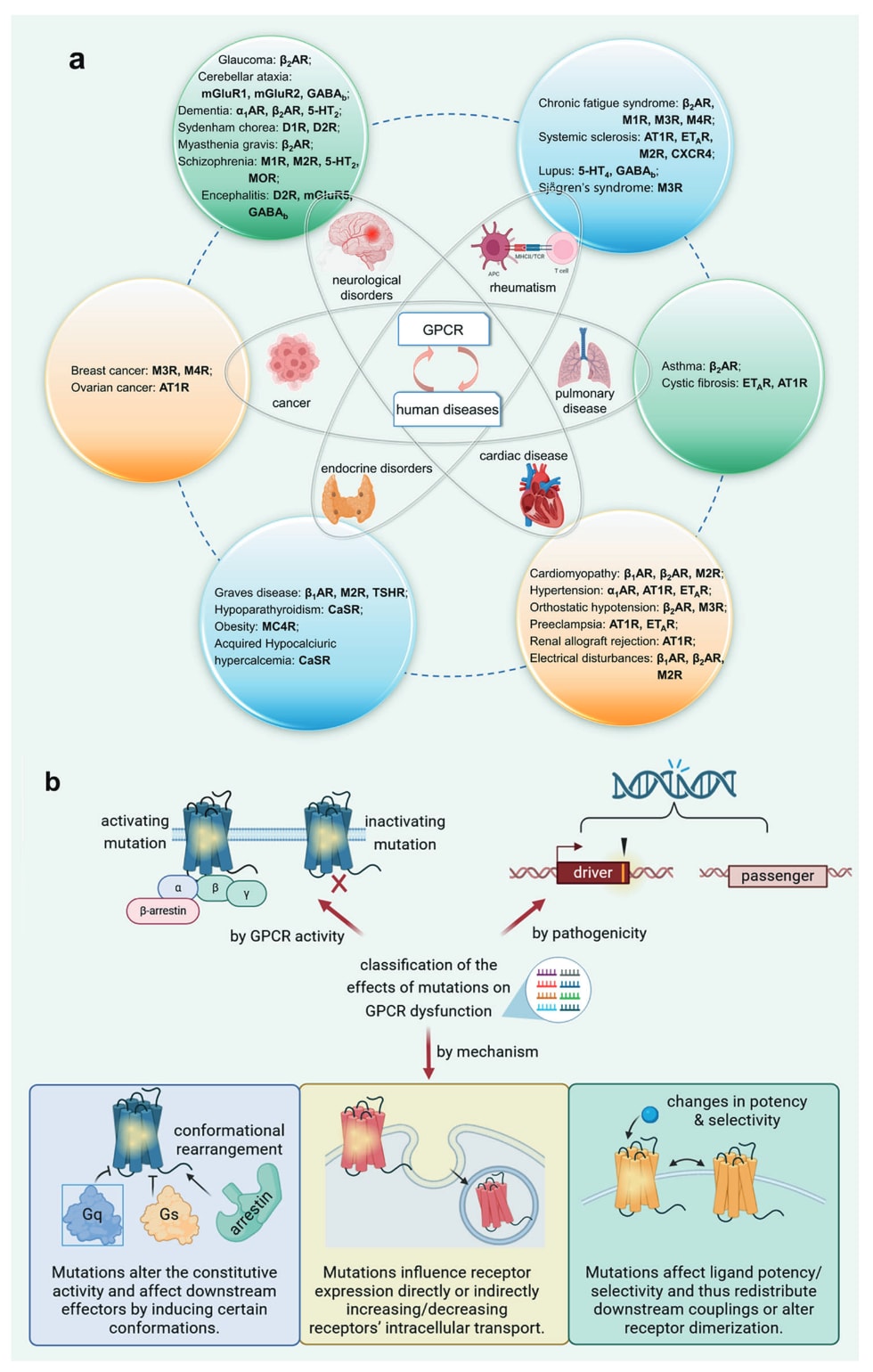
图4 a GPCR功能异常导致的人类疾病类型示例。b GPCR功能异常中突变作用的分类
5 GPCR药物发现的最新进展
5.1 GPCR药物发现的传统方法与新兴策略概述
自从脑啡肽被首次确认可作为阿片受体的内源性配体以来,具有多样调控作用的GPCR调节剂不断被发现,使GPCR研究持续获得新的意义。过去数十年间,GPCR药物发现领域从偶然性寻找转向理性设计,配体类型也从天然产物扩展到合成化合物与工程化抗体。当前除了传统的分子对接与结构基础药物设计外,多种湿实验筛选方法已被建立,用于更高效地筛选高质量先导化合物,如FRET/BRET、生物发光能量转移技术NanoBiT、Tango实验以及19F NMR等。当获得hits后,通常借助片段生长、性质预测与分子动力学模拟等计算方法,开展构效关系优化,从而启动从hit到先导,再从先导到候选药物的开发流程。
在此背景下,该综述特别强调合成小分子调节剂与GPCR结合时的相互作用模式与信号机制,希望为发现具有更高效力、更好选择性以及潜在偏向性效应的创新分子提供启发。
5.2 基于结构的GPCR正构位点药物设计
**以GPCR正构位点为靶点的结构基础药物设计中,小分子正构调节剂是最常见且最成熟的一类非肽类调控分子。**它们通过与内源性配体竞争结合正构结合口袋(OBP),引发受体内部结构的构象位移,从而发挥完全激动、部分激动或拮抗等作用。尽管正构小分子的开发相对成熟,但其亚型选择性低与信号多重耦联所带来的副作用依旧是核心难题。
**在过去三十年中,X射线与cryo-EM的广泛应用显著推动了GPCR−正构配体复合物的结构解析。目前已获得657个A类、16个B1类、6个B2类、19个C类、18个F类以及1个T类GPCR−正构配体结构(补充表1–5)。**在此基础上,文章精心挑选了近年获批的五个代表性复合物,用以解析配体识别机制、选择性来源以及精细的信号转导过程。此外还以两个案例展示结构信息在构效关系(SAR)优化以及结构−功能选择性关系(SFSR)构建中的关键作用。
通过对这七个案例的系统分析,文章旨在从已获批药物或高选择性分子中提炼关键信息,为未来开发高质量GPCR正构调节剂提供建设性思路,并为突破当前面临的选择性与安全性困境奠定基础。
5.2.1 与oliceridine复合的μOR结构
**吗啡与芬太尼(1)是治疗急性或慢性疼痛最有效的药物之一,其共同靶点μOR则同时介导镇痛作用与不良反应。**为了减轻副作用并扩大治疗窗口,能够抑制β-arrestin活性且保持相对完整G蛋白信号的调节剂一直是研发重点。Oliceridine(2)是一种结合于μOR正构位点的部分激动剂,于2020年获FDA批准,其优势在于能够通过偏向G蛋白通路传递信号,从而显著降低副作用(图5a、b)。因此解析oliceridine−μOR复合物的结构与偏向性信号的分子机制,对于开发新一代镇痛药具有重要意义。
通过比较μOR−oliceridine与μOR−fentanyl的复合结构可以发现,两者在Trp295^6.48之上的正构结合口袋内具有良好重叠的结合模式,唯一差别在于oliceridine的吡啶环相对芬太尼的n-苯胺基团向TM2倾斜约35°,导致其与TM6/7的疏水相互作用弱于芬太尼。根据Zhang等人的MD模拟结果,可推断更强的TM6/7相互作用能够促使TM6与TM7-H8向TM核心内移,形成同时适配G蛋白与β-arrestin的胞内口袋,因而产生中性信号;而相互作用较弱时,TM6/7胞内端更可能远离TM核心,从而稳定更偏向G蛋白结合与信号传递的胞内口袋。
基于这一机制,研究者设计了两种芬太尼衍生的μOR激动剂(3–4),通过将芬太尼中的苯胺基团替换为丙基或异丙基以降低其与TM6/7的疏水性,成功实现偏向G蛋白通路的信号传递(图5c)。区别于开发oliceridine过程中较为依赖“试错”的策略,Zhang等人的系统研究展示了如何通过解析复合结构来理解偏向信号的分子基础,并为利用SBDD设计具有偏向性的OR调节剂提供了有效范例。
5.2.2 与siponimod复合的S1PR结构
鞘氨醇-1-磷酸受体(S1PR)属于A类GPCR,共包含S1PR1至S1PR5五个亚型,调控淋巴细胞迁移、血管发育、内皮屏障稳定性与心率等多种生理功能。**尽管Fingolimod在2010年作为首个S1PR激动剂获得FDA批准,但其亚型选择性低,导致心动过缓与房室传导阻滞等多种“脱靶”效应。**因此,开发第二代、具有高亚型选择性的S1PR调节剂成为迫切需求。Siponimod(5)于2019年在全球获批,用于治疗成人复发型多发性硬化症,其选择性靶向S1PR1与S1PR5。深入理解其配体识别与受体活化机制对于解析亚型选择性及GPCR信号转导具有重要意义。
Yuan等人解析了siponimod−S1PR1−Gi以及siponimod−S1PR5的cryo-EM结构,显示配体以线性构象跨越正构口袋中极性模块与深部疏水腔(图6a)。由于S1PR家族在胞外前庭结构上存在差异,不同亚型具有不同的胞外脂层,这些差异构成配体进入受体的不同通道,从而影响亚型特异性(图6b)。进一步比较siponimod−S1PR1−Gi复合物与拮抗剂ML056结合的S1PR1结构,揭示受体活化过程中的“双重开关机制”。在配体结合后,Leu128^3.36向远离TM5方向旋转约130°,与siponimod的疏水部分形成直接相互作用,并破坏其与Trp269^6.48的原有相互作用,同时诱导Trp269^6.48协同向下移动。这两个关键残基的显著位移使TM3−TM6的相互作用松弛,进而引发TM6向外摆动,为G蛋白结合腾出空间(图6c、d)。这一活化机制与CB1与MC4R中对应的机械开关高度相似,由此提示通过设计能够与3.36位点形成精细疏水相互作用或直接促使3.36−6.48重新构象的配体,有望进一步提升活化效能。
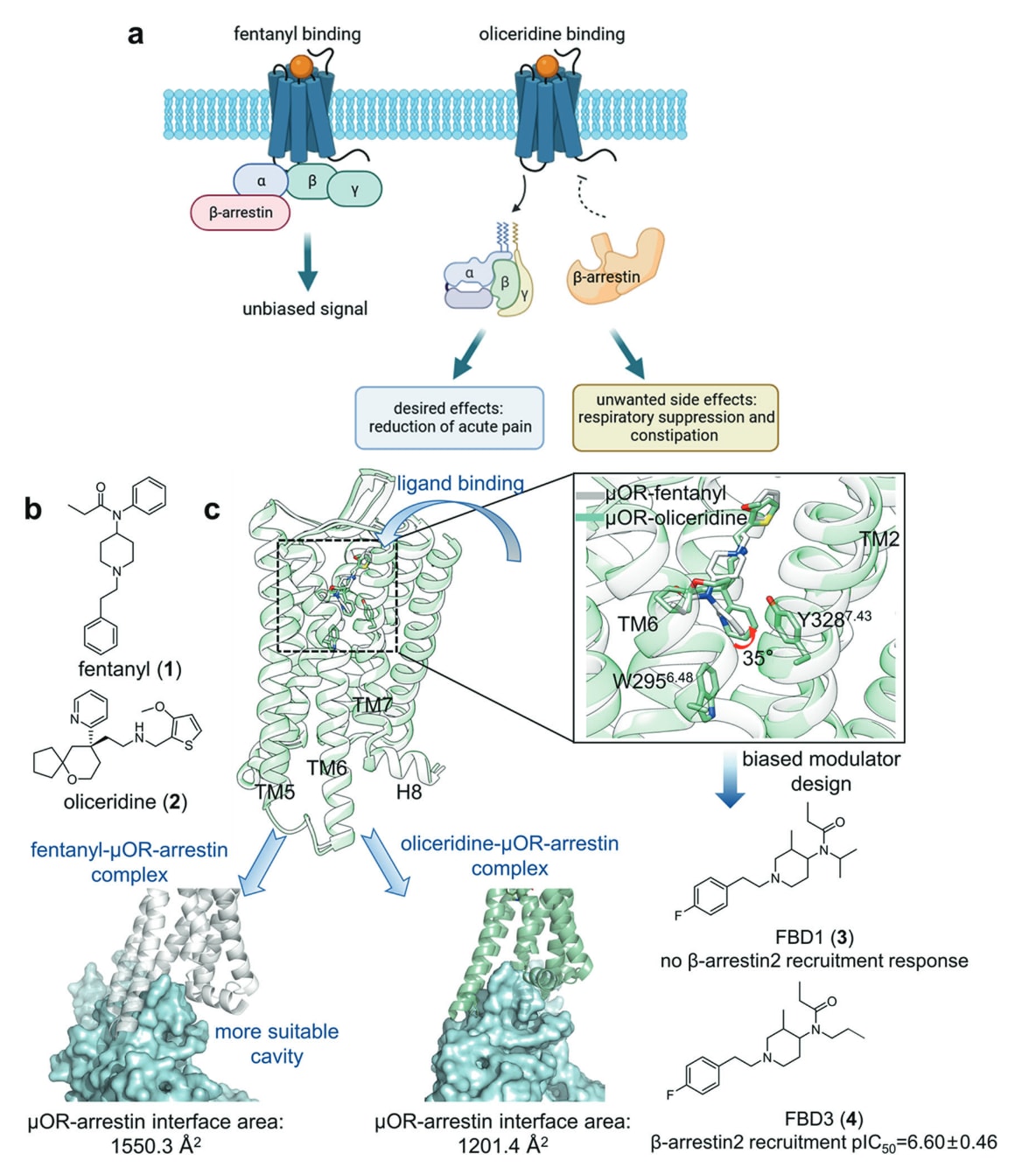
图5 a 芬太尼与oliceridine导致不同药理效应的整体示意。b 芬太尼与oliceridine的二维结构示图。c μOR−fentanyl(灰色)与μOR−oliceridine(浅绿色)复合物结构的叠合视图,并展示配体结合模式与arrestin耦联界面的比较,同时给出两种设计的偏向性调节剂的二维结构。
5.2.3 与lemborexant复合的OX2R结构
食欲素受体在中枢神经系统广泛表达,通过调控睡眠–觉醒节律展现出治疗失眠的潜力。OX1R与OX2R两个亚型分别主导不同的睡眠阶段,OX1R主要影响快速眼动(REM)睡眠,而OX2R则调控非快速眼动睡眠(NREM)与REM睡眠。Lemborexant(6)是一种正构竞争性拮抗剂,于2019年获FDA批准,对OXRs具有显著的抑制活性。然而,对lemborexant最关键的研究问题主要集中在两点:(1)为何其对OX2R相较OX1R具有中等水平的选择性,这对设计特异性调控OX1R/OX2R的分子以用于REM与NREM功能研究具有重要价值;(2)lemborexant的动力学参数基础为何,及其如何解释药物诱导的入睡改善与睡后觉醒时间减少之间的关系。
为阐明lemborexant的亚型选择性机制并为抗失眠药物开发提供指导,Asada等人解析了OX2R−lemborexant的晶体结构,并与此前已报道的OX1R−lemborexant结构进行比较。尽管配体均可与OX1R的Gln126^3.32及OX2R的Gln134^3.32形成相同的氢键,但由于OX1R中Ala127^3.33侧链体积较小,使lemborexant以两种取向结合;而在OX2R中,Thr135^3.33具有更大的体积,造成明显的空间阻碍,使配体仅能以单一构象结合。这被认为是造成其OX1R/OX2R亲和力差异的主要因素(图7a、b)。
进一步通过模拟研究发现,lemborexant在溶液中的两个芳环之间存在分子内堆叠作用,使其自由态构象更接近于结合态,从而解释其较高的kon值。此外,与其他OXRs调节剂相比,lemborexant具有更高的结合自由能,这或许与其较高的koff有关。整体来看,通过分子内相互作用优化自由分子的构象以提升kon是可行策略(图7c、d)。扩展而言,分别调控配体结合受体时的焓贡献与自由分子内部结构带来的熵效应,或将成为设计具有更好动力学特征与动态行为药物的重要方向。
5.2.4 与lasmiditan复合的5-HT1F结构
5-HT1受体家族包括5-HT1A、5-HT1B、5-HT1D、5-HT1E与5-HT1F,属于典型的A类GPCR,能够响应内源性神经递质5-羟色胺,被证实是治疗偏头痛、抑郁与精神分裂症的潜在靶点。尽管传统的靶向激动剂已用于偏头痛治疗多年,但由于其对5-HT1B与5-HT1D的非选择性激活而引起血管收缩等副作用,始终是临床应用中的重大限制。Lasmiditan(7)因其高选择性与强效的5-HT1F激动特性,并具备良好中枢通透性,于2019年获FDA批准,用于避免传统药物的血管收缩风险。解析lasmiditan的骨架特征以及5-HT1F选择性活化机制,对于设计更安全的抗偏头痛药物十分关键。
Huang等人解析了5-HT1F−lasmiditan−Gi1复合物结构,呈现lasmiditan的整体结合模式。在正构结合口袋中,配体上甲基哌啶基团的伯胺通过与Asp103^3.32形成经典的电荷互作,并同时与Tyr337^7.42形成氢键,从而大幅稳定配体。在其延展结合口袋(EBP)中,lasmiditan的三氟苯基团通过疏水作用与Ile174^ECL2和Pro158^4.60相互作用,并与Glu313^6.55、Asn317^6.59、Thr182^5.40和His176^ECL2形成额外氢键。将5-HT1F与其他四个5-HT1亚型进行结构对齐后发现,其他亚型高度保守的TM4−TM5−ECL2区域在5-HT1F中发生显著构象变化,使lasmiditan无法在这些亚型中形成相同关键相互作用。因此,设计能够适配EBP并与TM4−TM5−ECL2区域产生特异作用的配体,有望实现高选择性5-HT1F激动(图8a、b)。
活化机制分析显示,lasmiditan会诱导关键开关残基Trp^6.48向下移动,并进一步引发PIF、DRY与NPxxY基序的构象重排。结构比较还表明,5-HT1F−Gi复合物中的αN结构相对于其他5-HT1受体−Gi/o复合物有明显偏移,说明其具有独特的Gi耦联方式与相应的特异下游效应(图8c)。因此,通过设计能够与关键开关残基互作并兼具优化血脑屏障通透性的分子,有望获得更高效、更安全的5-HT1F激动剂。
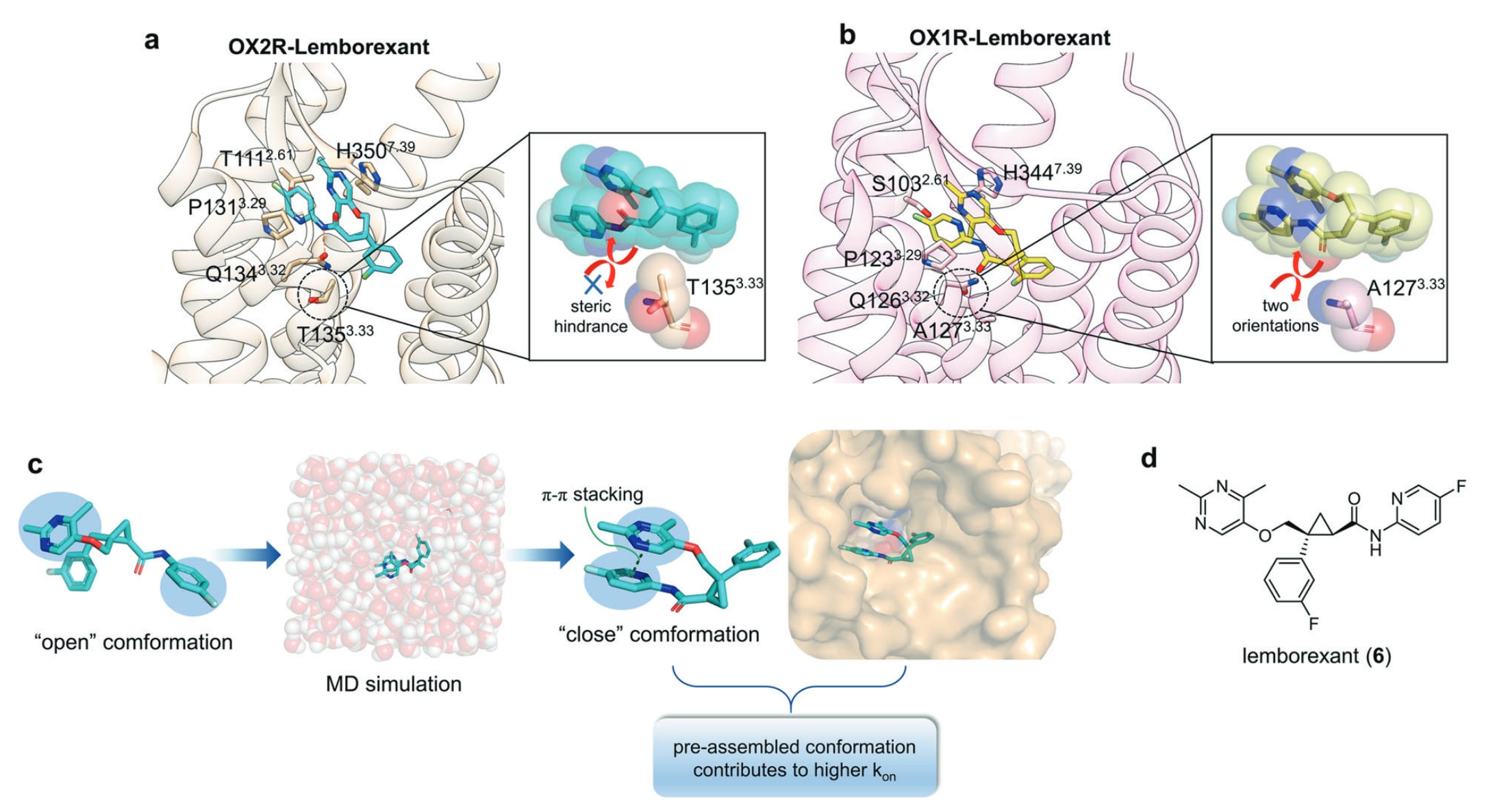
图7 a OX2R−lemborexant复合物的详细结合模式(受体:浅橙色,配体:青色,PDB: 7XRR),其中Thr135^3.33的空间阻碍仅允许配体以单一取向结合。b OX1R−lemborexant复合物的结合模式(受体:浅粉色,配体:黄色,PDB: 6TOT),其中Ala127^3.33侧链较小,使配体能够以两种取向结合。c 通过MD模拟预测配体在与受体结合前的构象,以提升kon值的示意图。d lemborexant的二维结构示意图。
5.2.5 与elagolix复合的GnRH1结构
典型的A类GPCR——促性腺激素释放激素受体1(GnRH1R),在受到其内源性肽类激动剂GnRH激活后,会通过Gq蛋白通路启动生殖激素级联反应并促使促性腺激素的释放。随着GnRH1R首个非肽类拮抗剂elagolix(8)于2018年上市,人们对GnRH1R−elagolix复合物的结构特征产生了浓厚的药理学兴趣。此外,与其他A类GPCR不同,GnRH1R在胞质侧缺少C端螺旋(helix 8),并在2.50位置上具有天冬酰胺而非其他受体普遍保守的天冬氨酸,这为其7TM结构域内信号级联过程中的微开关模式带来了更大的可塑空间。
Yan等人解析的GnRH1R−elagolix晶体结构显示,由Lys121^3.32与Asp98^2.61组成的极性网络在与配体形成极性相互作用中起关键作用,而Tyr283^6.51与Tyr290^6.58则通过疏水相互作用参与配体识别(图9a)。值得注意的是,elagolix结合位置更靠近TM7,使正构结合口袋扩大,从而允许该位点由N端进入并产生共同占位现象。
结构对齐与IP累积实验进一步表明,与部分GPCR可由配体直接作用于Trp^6.48触发toggle switch不同,GnRH1R的TM6中Tyr283^6.51–Tyr284^6.52–Trp280^6.48–Phe276^6.44构成的特殊基序被认为是介导信号传递的重要结构模块(图9b)。此外,仅约4%的A类GPCR在5.58位置为天冬酰胺,GnRH1R便是其中之一,该残基能够与Ser136^3.47形成极性作用,使TM6与TM3和TM5紧密堆叠,从而维持拮抗状态。
总体来看,这些发现揭示了GnRH1R在与代表性拮抗剂结合时的独特结构特征,也为结构生物学与药物设计提供了新的启示。
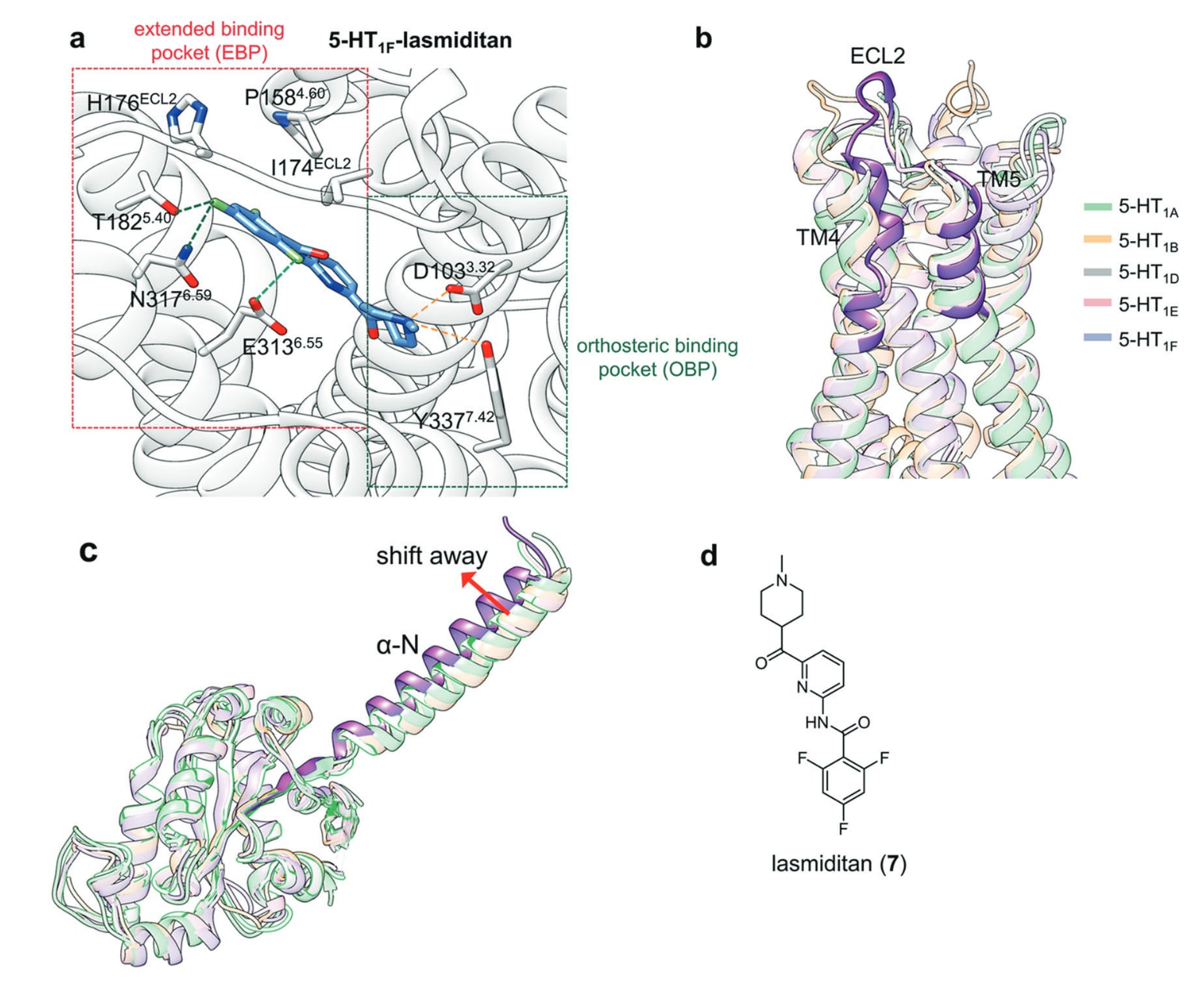
图8 a 5-HT1F−lasmiditan复合物的详细结合模式。氢键以橙色虚线表示,卤键以绿色虚线表示。b 5-HT1A(浅绿色,PDB: 7E2X)、5-HT1B(浅橙色,PDB: 5V54)、5-HT1D(浅灰色,PDB: 7E32)、5-HT1E(浅粉色,PDB: 7E33)与5-HT1F(浅紫色,PDB: 7EXD)的结构叠合图,突出显示5-HT1F受体的TM4−ECL2−TM5区域。c G蛋白与相应5-HT受体结合时αN螺旋的结构比对,其中5-HT1F−Gi复合物的αN螺旋被特别标示。d lasmiditan的二维结构示意图。
5.2.6 利用结构信息开展SFSR研究
过去十年中,晶体学的发展展示了基于结构的药物设计(SBDD)在GPCR研发中的巨大潜力,但目前主流策略仍以构效关系(SAR)研究与基于片段的药物设计(FBDD)为主。这在一定程度上源于“活性断崖”现象,即微小结构变化会导致药效急剧改变,削弱了结构信息的利用价值。然而,结构驱动的研究前景依旧值得期待。以下两个案例展示了利用结构信息开展结构−功能选择性关系(SFSR)研究的潜力。
第一个例子是通过SBDD策略高效研发并优化A2AR选择性拮抗剂1,2,4-三嗪衍生物4d(9)。通过生物物理映射(BPM)与晶体结构分析可知,4d主要通过三嗪核心与N253^6.55形成的两个氢键稳定结合,其A环朝向TM2与TM7(图10a)。基于此,研究者提出在A环对位引入氢键受体以与His278^7.43互作,并在同一芳环引入疏水取代基与Ile66^2.64作用作为SAR优化方向。通过在A环对位加入苯酚羟基或4-吡啶氮,并结合其他小体积脂溶性取代基进行亲和力微调,成功获得效能最佳的4k(图10b)。进一步比较A2AR分别与腺苷(激动剂)、ZM241385(拮抗剂)以及4e(拮抗剂)的结合口袋可知,腺苷类激动剂会以核糖环填充下部腔体的疏水亚口袋,而拮抗剂结合时该区域通常空置。此外,该区域亦可用于提升A2AR相对A1AR的选择性(图10c)。因此,将新化学骨架拓展至该区域有望进一步提升功能多样性与选择性。
第二个案例展示了以aripiprazole为起点,通过结构指导设计偏向β-arrestin的D2R激动剂,以降低抗精神病药物常见的运动障碍副作用。研究者首先将aripiprazole的二氯苯基哌嗪部分替换为吲哚哌嗪,得到化合物12,其在Gi/o介导的cAMP抑制与β-arrestin2募集实验中表现出相当活性。分子对接显示12的吲哚NH可与Ser^5.42形成氢键,而此残基已知在β2肾上腺素受体中介导G蛋白信号,因此研究者在吲哚NH上引入甲基,以调整结合构象并避免TM5参与(图11a)。参考同源5-HT2B受体结构中配体与ECL2疏水残基的关键互作,研究者进一步在吲哚2位加入第二个甲基,得到偏向β-arrestin的化合物13,其偏向因子达20,并可能减少副作用(图11b)。据目前资料,这是首次利用结构信息理性设计GPCR偏向性配体的成功案例,突显了在SFSR研究中进行结构比较的必要性。

图9 a GnRH1R−elagolix复合物的详细结合模式(受体:浅灰色,配体:浅粉色,PDB: 7BR3),其中GnRH1R的N端以浅紫色标示,用以展示其与elagolix在正构结合口袋中的共同占位。b GnRH1R中特殊信号传导机制的概览示意。c elagolix的二维结构示意图。
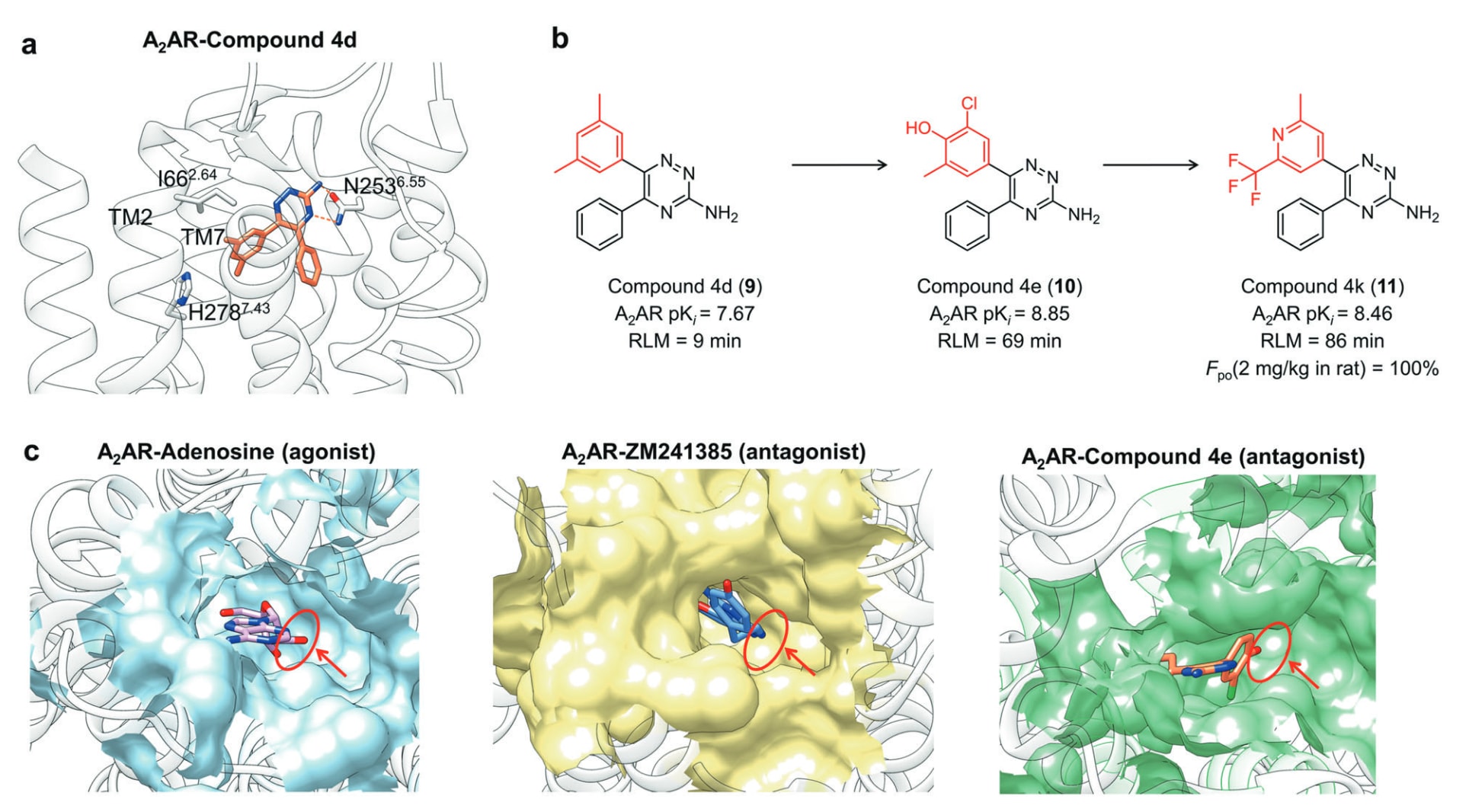
图10 a A2AR−4d复合物的详细结合模式(受体:浅灰色,配体:橙色,PDB: 3UZA)。b A2AR拮抗剂的构效关系研究。c A2AR−腺苷复合物(浅蓝色,PDB: 2YDO)、A2AR−ZM241385复合物(浅黄色,PDB: 4EIY)与A2AR−4e复合物(浅绿色,PDB: 3UZC)的正构结合位点对比,其中腔体占位差异以红色圆圈与箭头标示。
5.3 解析与小分子变构调节剂复合的GPCR结构及其变构信号机制
过去十年,随着对GPCR变构调控认识的不断深化,GPCR靶向变构药物的研发在结构层面取得了显著进展。截至2024年2月,已有59个与GPCR结合的小分子变构调节剂的结构被解析,包括33个A类、7个B类、18个C类以及1个F类受体的复合结构。尽管GPCR本身具有高度动态性且不同受体间结构多样,但目前发现可作为变构口袋的位置十分有限,并且这些口袋在不同同源程度的GPCR之间也具有一定共性。甚至在一个受体内部,也可发现多个变构位点。
可成药的变构热点遍布整个受体,可大致划分为胞外前庭、跨膜结构域、胞内表面、7TM结构域外侧以及7TM结构域内部数个区域。目前已知所有GPCR中的变构结合位点可归为11种不同位置,并具有某些共有特征(图12)。图中将各受体上发现的不同变构口袋映射到一个示例GPCR结构上,便于它们之间的对比。
根据变构配体对正构配体刺激活性的影响方式,可将变构配体分为正向变构调节剂(PAM)、负向变构调节剂(NAM)、变构调节剂以及变构反向激动剂。PAM如Cinacalcet,可增强钙敏感受体(CaSR)对正构激动剂的响应;NAM如Mavoglurant,可削弱mGluR5对正构激动剂的反应。Ago变构调节剂能够在无正构激动剂存在时直接激活或抑制受体,例如靶向GLP-1R的化合物2。
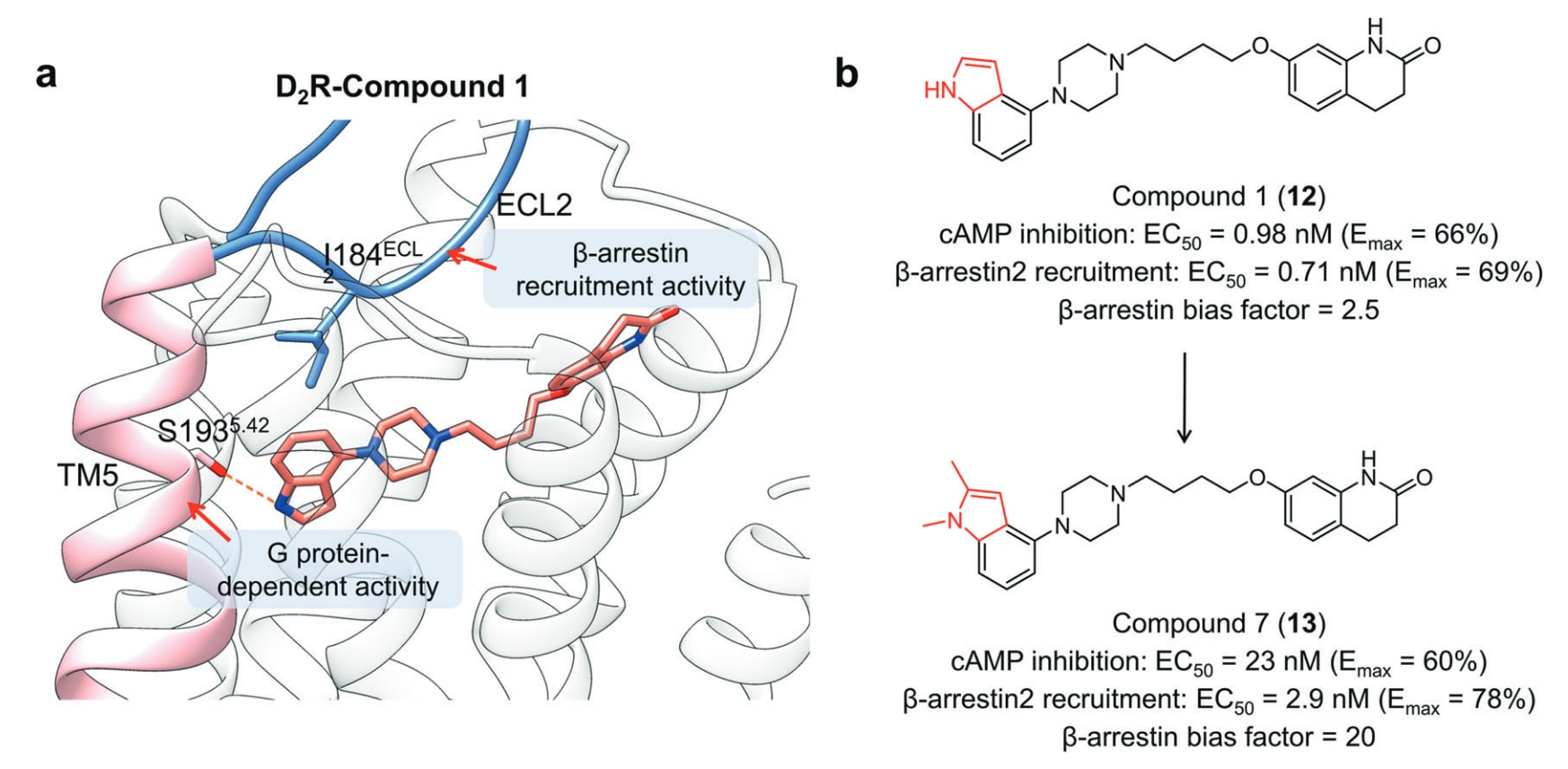
图11 a D2R−化合物1(12)复合物的详细结合模式(受体:浅灰色,配体:鲑红色,受体基于PDB: 3PBL构建)。其中受体的TM5以粉色标示,ECL2以蓝色标示以便观察。b D2R β-arrestin偏向性激动剂的构效关系研究。
表1|与合成变构调节剂结合于胞外前庭区域的GPCR已解析结构
自2004年首个钙敏感受体(CaSR)正向变构调节剂PAM——cinacalcet获FDA批准用于治疗甲状旁腺功能亢进以来,结合于GPCR胞外前庭的小分子变构调节剂发展迅速。迄今已有四种此类调节剂获批,另有一种进入临床试验,具体情况汇总于表1。已解析的晶体结构显示,胞外前庭上存在三个主要结合位点,分别位于I−II螺旋外侧、II−III螺旋外侧以及7TMD内部(图13)。由于这些变构位点毗邻A类与B类GPCR的传统活性位点,因此其调节作用往往可通过直接改变正构配体与受体的结合方式来实现。鉴于GPCR具有共同的进化祖先,这一区域的变构口袋也被认为可能代表了受体的“祖先正构位点”。

5.3.1 靶向GPCR胞外前庭区域(7TMD外侧与内侧)
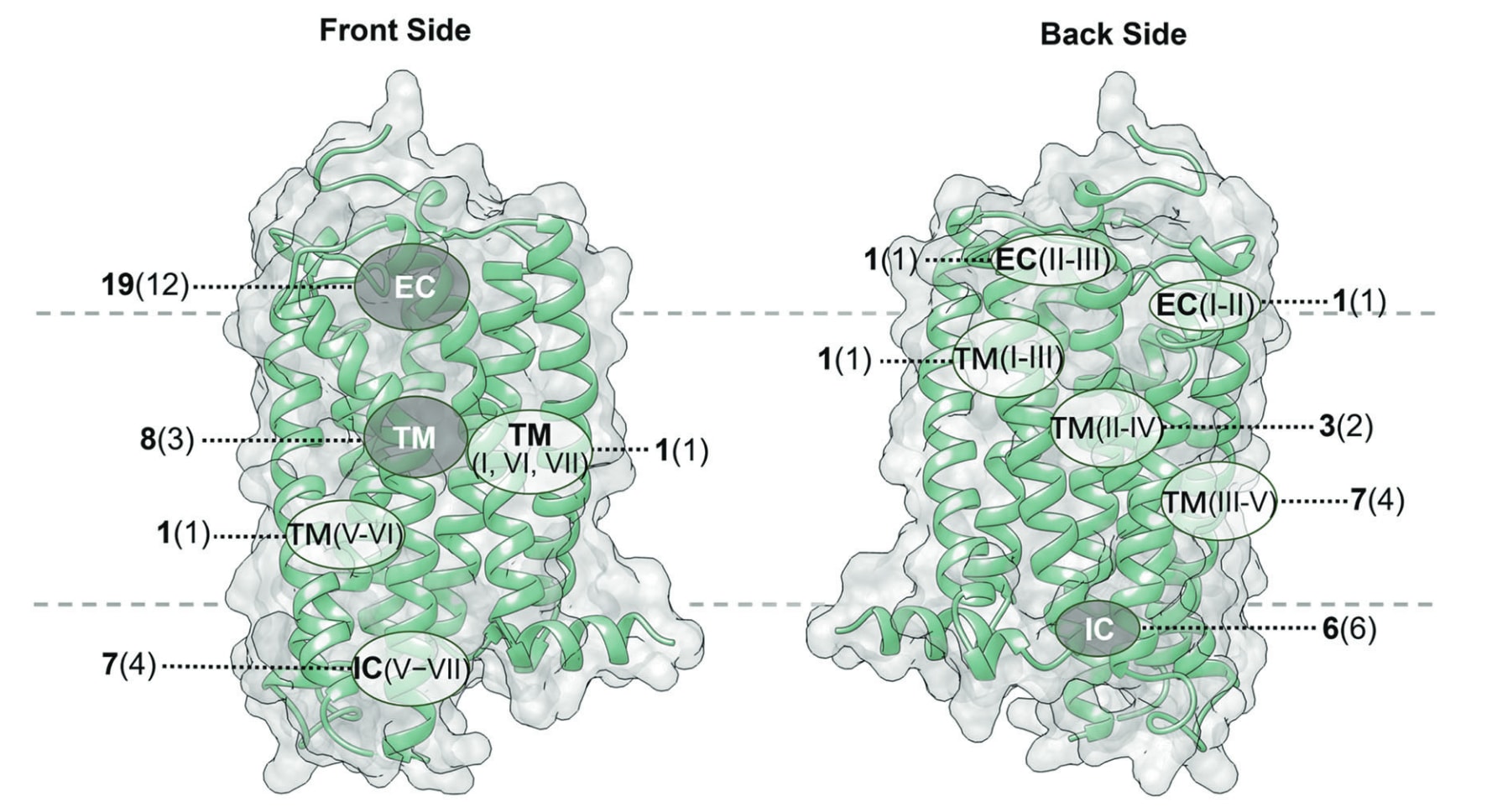
图12|在GPCR中已报道的11个变构结合位点映射至代表性A类GPCR CB1R结构。 灰色区域表示7TMD内部的结合口袋,白色区域表示7TMD外侧的结合口袋。每个口袋旁以黑体标注该口袋中已知的独特配体数量,并在括号中标明含有该口袋的GPCR数量。灰色虚线表示脂双层的边界。

图13|GPCR胞外区域的三类变构结合位点及其对应的小分子变构调节剂。 小分子配体以棒状模型映射到三个代表性受体上:7TMD外侧(I−II)的GLP-1R(PDB: 6VCB)、7TMD外侧(II−III)的GPR101(PDB: 8W8S)以及7TMD内部的M4R(PDB: 7V68)。为便于比较,将M4R正构配体的位置以灰色球棒模型标示于整体图中。每个口袋旁以黑体标注其已知的独特变构调节剂数量,并在括号中标注含有该口袋的GPCR数量。
- 7TMD外侧(I−II):GLP-1R−LSN3160440结构
胰高血糖素样肽1受体(GLP-1R)属于B类肽激素GPCR,其活化能够在葡萄糖依赖的条件下促进胰岛素分泌并减少胰高血糖素的释放。对于此类肽类受体而言,相比正构位点,小分子更容易靶向其变构口袋,因此开发高效的GLP-1R激动剂与PAM对于2型糖尿病治疗至关重要。
LSN3160440(14)(图14)是一种GLP-1R的小分子PAM,EC50约1 μM,可增强GLP-1(9–36)的效力与效应,使其成为完全激动剂。GLP-1R−LSN3160440−GLP-1−Gs的cryo-EM结构清晰展示了LSN3160440的U形结合模式,其变构结合位点由胞外前庭中的TM I与TM II残基共同构成(图15a)。
在结合口袋中,LSN3160440的苯并咪唑结构(图15c)与Leu142^1.37形成疏水互作,并与Tyr145^1.40形成芳香堆叠(图15b)。突变与MD模拟也提示,苯并咪唑N3可能通过水分子介导与Lys202^2.72形成氢键。值得注意的是,LSN3160440还能与GLP-1同时形成相互作用,发挥类似“分子胶”效应,其2,6-二氯-3-甲氧基苯基部分可同时与GLP-1的Phe12、Val16与Leu20产生疏水接触。
目前已有多种GLP-1R的小分子正构激动剂复合结构被解析(图15d)。这些配体与LSN3160440的结构对比显示,它们在TM1−TM2裂隙的胞外端共享一个关键区域,提示该区域对于正构与变构激动剂的先导优化均具有潜在价值。此外,在结合位点中,与Tyr145^1.40的芳香相互作用是保守特征。
- 7TMD外侧(TM I–II):GPR101−AA-14结构
GPR101是一类孤儿A类GPCR,在伏隔核与下丘脑中高度表达,具有固有的Gs与Gq活性。GPR101的基因重复或突变会调节其固有活性,使其成为代谢疾病颇具吸引力的治疗靶点。近期研究发现AA-14(15)(图14)是GPR101的变构激动剂,能够强效激活Gs并具备极高的亚型选择性。动物实验显示,AA-14可通过激活垂体GPR101发挥抗衰老作用。AA-14−GPR101−Gs复合物的cryo-EM结构揭示AA-14在受体上具有两个独立结合位点(图16a),其一位于7TMD外侧,被TM I、TM VI与TM VII环绕;另一位于TM2−TM3及ECL1外侧。在胞外变构口袋中,AA-14的3-三氟甲基苯基(图16c)与Asn100^ECL1形成极性交互,并与Phe103^3.24和Trp87^2.60形成疏水作用(图16b),而其4-甲基-2-吡啶基则堆叠于Phe96^ECL1与Leu99^ECL1之上。
- 7TMD内部变构位点:多个GPCR复合结构
这一类变构口袋位于7TM螺旋束形成的胞外腔体内,位置正处于A类与B类GPCR正构位点以及SMO胆固醇结合位点之上(图13)。这是目前药物样变构调节剂最常靶向的变构位点,因为小分子可从胞外进入而无需穿膜。该区域通常可分为正构子口袋和变构子口袋两部分,其大小受N端区和ECL2调控,从而为多样化配体创造新的结合空间。
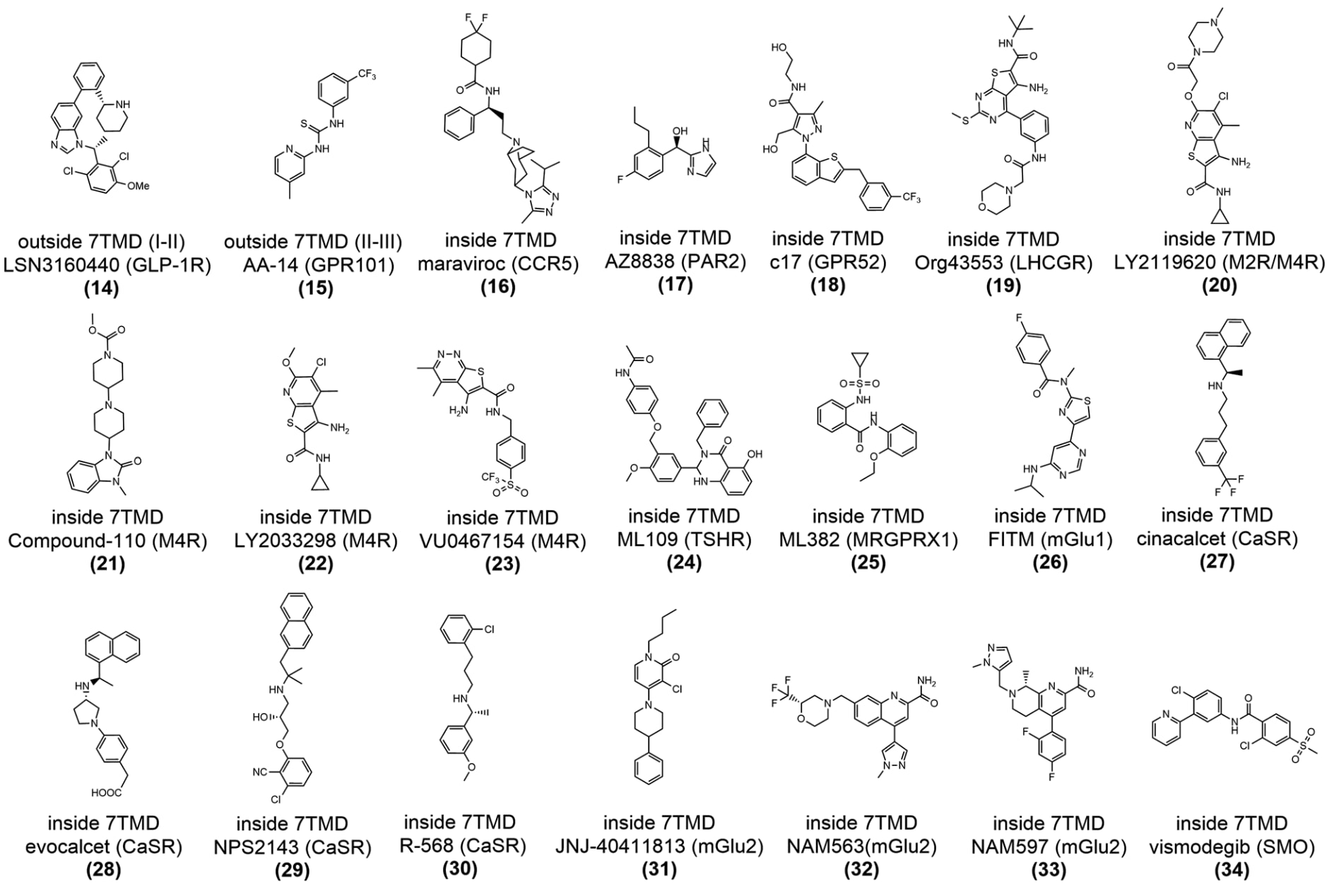
图14|靶向GPCR胞外前庭区域的合成变构配体的二维化学结构示意图
5.3.2 以M2R/M4R−LY2119620为典型例
毒蕈碱受体(mAChRs)M1–M5是A类GPCR的重要代表,调控中枢与外周神经系统的乙酰胆碱释放,与阿尔茨海默病、精神分裂症与成瘾等疾病密切相关。LY2119620(20)(图14)是M2R与M4R的PAM,但因交叉反应与潜在心血管风险而不适用于治疗。M2R与M4R−LY2119620的结构均已解析。虽然其结合模式高度相似,但仍存在细微差异(图17a)。LY2119620结合于正构位点上方的宽敞胞外前庭,与正构口袋由Tyr^3.33、Tyr^6.51与Tyr^7.39隔开。在M2R中,LY2119620的噻吩并吡啶环被Tyr177^ECL2与Trp422^7.35夹持(图17b),而在M4R中,则被Phe186^ECL2与Trp435^7.35夹持(图17c)。此外,M2R中的Tyr80^2.61、Asn410^6.58与Asn419^ECL3可与配体形成氢键,而M4R则仅有Gln427^ECL3参与氢键作用。
MD模拟显示,LY2119620可调节Trp422^7.35构象,继而驱动Tyr426^7.39在正构口袋中的重新定位,这可能解释其对正构激动剂亲和力提升的机制(图17d)。
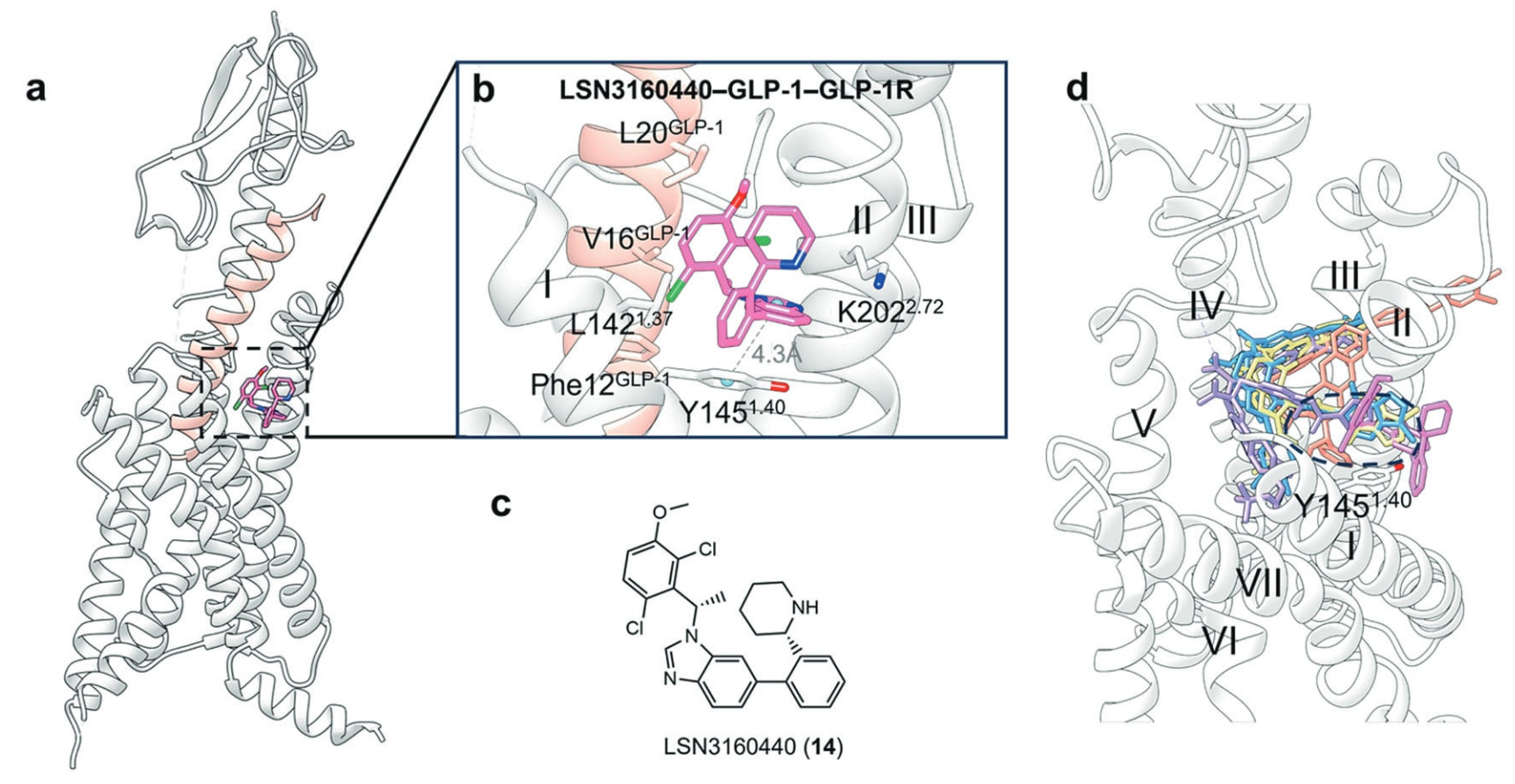
图15 a GLP-1R同时结合PAM LSN3160440与正构配体GLP-1的示意图(PDB: 6VCB),其中GLP-1以粉色标示。b GLP-1R−LSN3160440复合物的详细结合模式,π–π堆叠以灰色虚线表示。c 小分子变构配体LSN3160440的二维结构示意。d 将正构小分子激动剂Boc5(紫色棒状)、TT-OAD2(鲑红色棒状)、LY3502970(黄色棒状)、CHU-128(蓝色棒状)叠合至LSN3160440−GLP-1−GLP-1R结构,可观察到其在TM1−TM2裂隙处的部分重叠,并突出显示保守残基Tyr145^1.40。
5.3.3 以CaSR变构调节剂为例
CaSR属于C类GPCR,是控制钙稳态的关键受体。Cinacalcet(27)(图14)是其PAM,用于继发性甲旁亢治疗;而calcilytic NPS-2143(29)是其NAM。两者在CaSR中的结合姿态明显不同。
Cinacalcet可呈“延展”或“弯折”两种构象,与CaSR形成稳定复合体(图18a)。其萘乙胺片段与Ile777^5.44形成疏水作用,并与Phe684^3.36和Trp818^6.50形成边对面π–π作用,牢固固定Trp818^6.50于7TM内部。不同构象下其苯基位置牵动Tyr825^6.57构象变化(图18b、c)。
相比之下,NPS-2143与CaSR形成稳定的“新月形”构象(图19a)。其末端萘基被Phe684^3.36、Leu776^5.43、Ile777^5.44、Trp818^6.50与Ile841^7.37包围(图19b),其3-氯-2-氰基苯基伸向侧开口并与Leu773^5.40及Tyr825^6.57相互作用(图19c)。
CaSR−NPS-2143结构在有无Ca^2+条件下高度一致,但Cinacalcet结合则显著改变Trp818^6.50、Phe821^6.53与Tyr825^6.57构象(图19d),促使TM6弯曲并稳定二聚界面,从而激活受体。相反,NPS-2143增强TM6螺旋性,抑制受体活化。
基于Cinacalcet的特殊结合特征,Liu等研究者对12亿化合物进行大规模虚拟筛选,通过采样“延展”与“弯折”两种构象空间,总共得到682万亿种构象组合,最终获得3.8%与13.6%的命中率,并优化得到高效候选物36(图19e)。这一策略展示了利用已解析结构与构象采样进行变构药物发现的范式,也具有推广潜力。
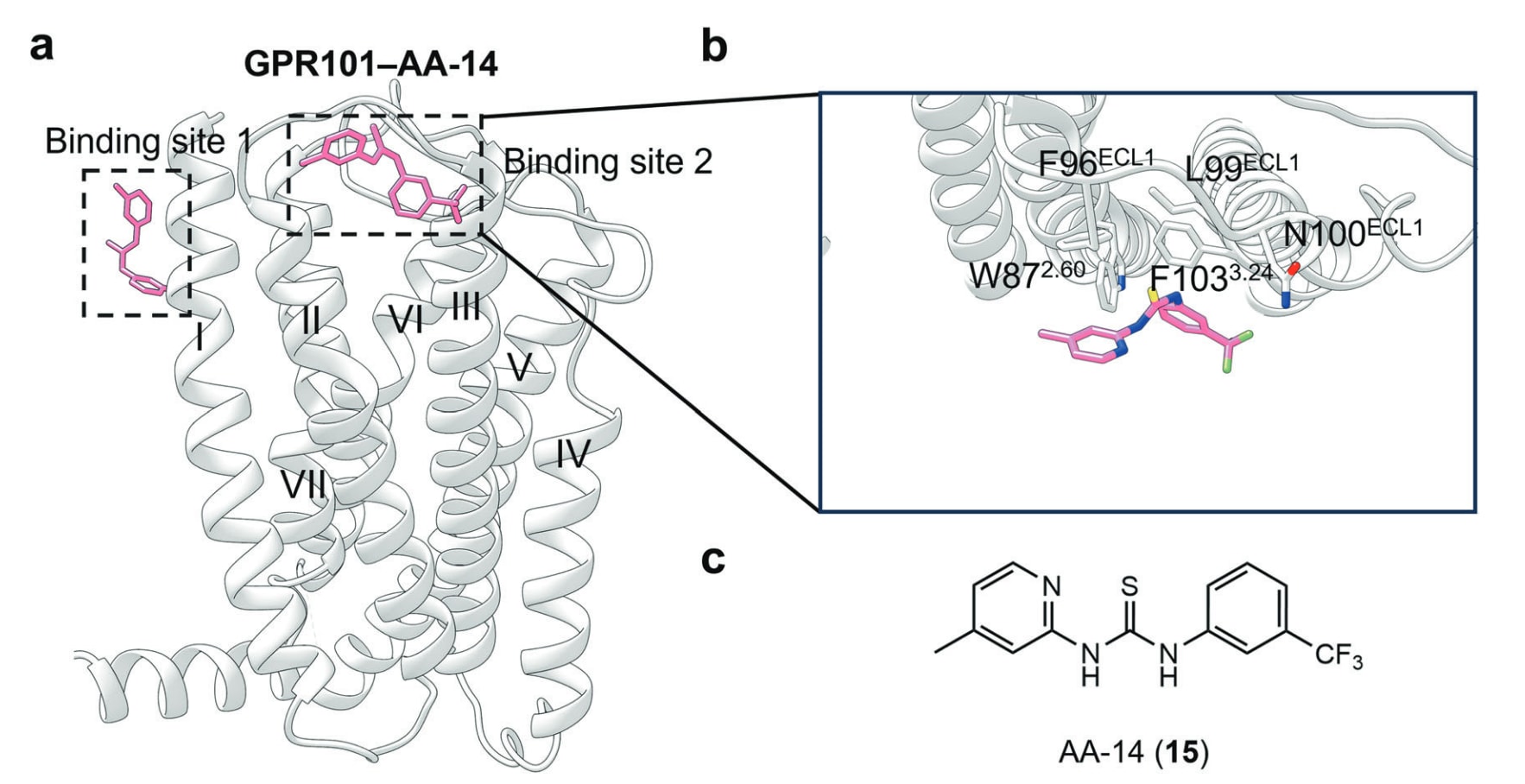
图16 a GPR101结合变构激动剂AA-14的示意图(PDB: 6VCB)。b GPR101−AA-14复合物的详细结合模式。c 小分子变构配体AA-14的二维结构示意图。
5.3.4 靶向GPCR跨膜结构域(7TMD外侧)
从已解析的结构来看,GPCR可利用位于7TMD外侧、紧邻脂质界面的跨膜结构域区域来结合变构调节剂。迄今为止,已有五类位于7TMD外侧的跨膜变构位点被结构学明确界定(Table 2),分别位于:I–III螺旋外侧、II–IV螺旋外侧、III–V螺旋外侧、V–VI螺旋外侧,以及I、VI、VII螺旋外侧(图20a、c)。这些位点主要出现在A类GPCR中。
由于这些口袋较浅,且不像传统正构位点那样被7TM螺旋束完整包围,因此在此类位点结合的变构调节剂通常携带若干极性基团,这些极性基团可与暴露在受体−脂双层界面的氢键供体或受体残基形成锚定作用。同时,这些配体也需要保持整体疏水性质,以便顺利进入跨膜区域。
结合于跨膜结构域(7TMD外侧)的变构调节剂通常通过稳定受体外侧的活性或非活性相互作用网络、或阻碍或促进受体活化过程中必需的跨螺旋运动,从而调节GPCR信号转导。换言之,它们从7TM螺旋束外部调控受体功能,呈现出不同于传统正构调节的独特机制。
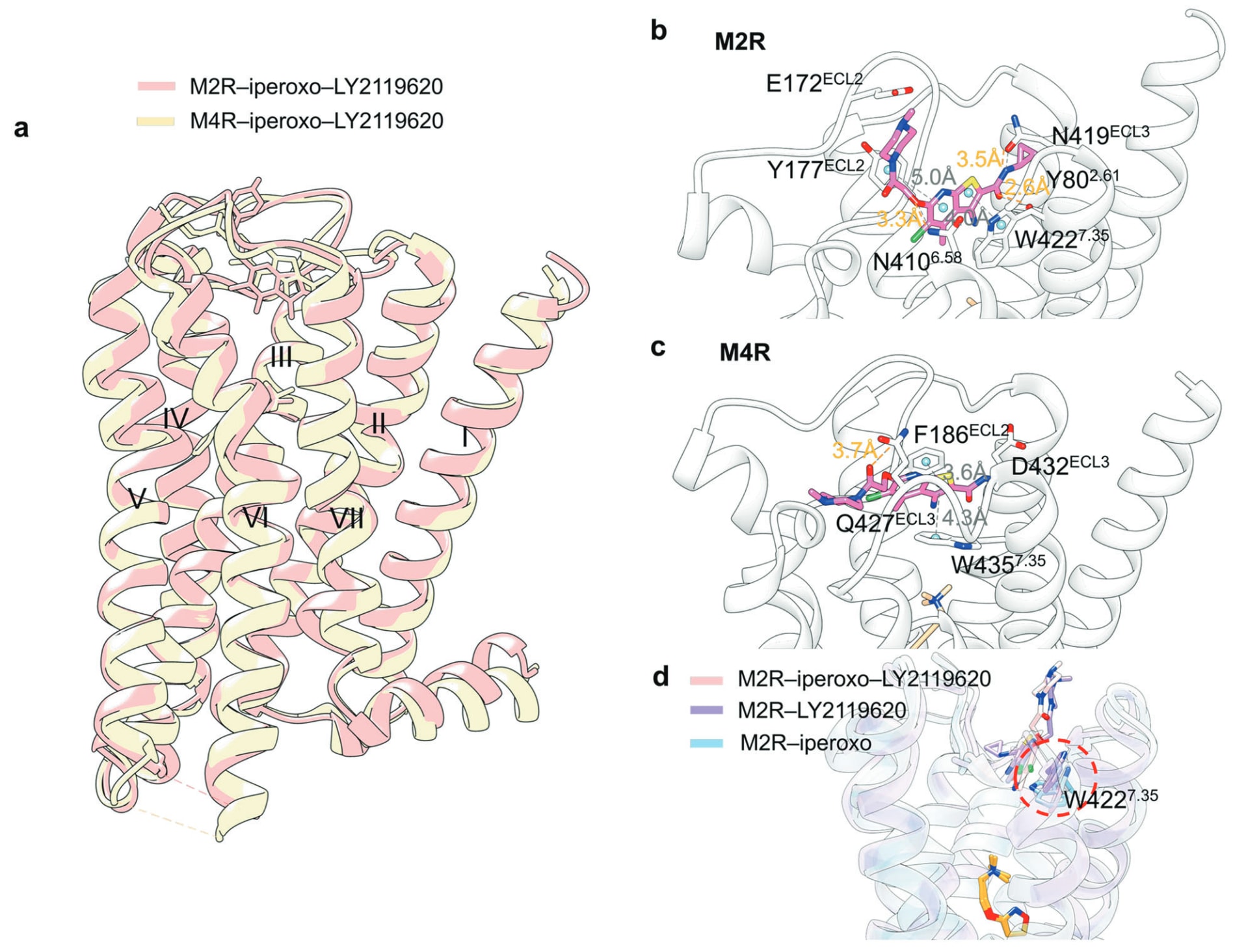
图17 a PAM LY2119620分别结合于M2受体(粉色蛋白、粉色配体;PDB: 4MQT)与M4受体(黄色蛋白、黄色配体;PDB: 7V68)的叠合视图。b M2受体−LY2119620复合物的详细结合模式。c M4受体−LY2119620复合物的详细结合模式,氢键以橙色虚线表示,π–π堆叠以灰色虚线表示。d M2受体中关键残基Trp422^7.36在三种结构中的叠合视图,包括M2受体−iperoxo−LY2119620(粉色;PDB: 6U1N)、M2受体−LY2119620(紫色;PDB: 4MQT)以及M2受体−iperoxo(蓝色;PDB: 4MQS),其中正构激动剂iperoxo以橙色标示。
- TM I–III:P2Y1−BPTU结构
迄今为止,仅有极少数小分子能够变构性地靶向该区域相对浅表的口袋。由于TM螺旋束表面较平坦、口袋狭窄,在此区域开展理性药物设计具有明显挑战。在已解析的案例中,P2Y1嘌呤能受体便是代表性靶点。P2Y1受体的激动会促进血小板聚集并触发血小板分泌,因此其拮抗剂被视为治疗血栓的潜在药物。
BPTU(37)(图20b)是一种P2Y1拮抗剂,也是首个结合于螺旋束外部的GPCR小分子变构调节剂,能够以纳摩尔级效力抑制P2Y1诱导的血小板聚集,且对P2Y1具有良好选择性(P2Y1 Ki = 75 nM,P2Y12 Ki > 70 μM)。P2Y1−BPTU二元复合结构显示,其结合口袋主要由TM I–III的残基构成(图21a)。
在极性相互作用中,BPTU脲基上的两个NH分别与Leu102^2.55的主链羧基形成关键氢键(图21b)。在疏水作用方面,BPTU的吡啶基与Ala106^2.59及Phe119^ECL1接触;其叔丁基苯环嵌入由TM II与TM III共同构成的疏水亚口袋,与Leu102^2.55、Thr103^2.56、Met123^3.24、Leu126^3.27和Gln127^3.28等残基相互作用。相对的一端,三氟甲氧基苯基与Phe62^1.43及Phe66^1.47形成疏水接触。
这一结构案例突显了变构配体的“形状互补”机制:7TM螺旋上的结构突起可作为锚点固定配体,如P2Y1中的Pro105^2.58便是此类锚点,并在74%的非嗅觉A类GPCR中保守存在。
将P2Y1结合激动剂2MeSADP与结合变构拮抗剂BPTU的结构进行比较可知,BPTU促使Tyr100^2.53向TM3方向偏移1.4 Å(图21c),从而阻止Phe131^3.32的构象变化。随后,Phe131^3.32与Phe276^6.51相互作用,限制TM6的移动,而TM6的外摆正是A类GPCR活化的关键步骤。
此外,MD模拟显示,BPTU结合后可稳定螺旋束,提升脂质层有序性,并进一步稳定失活构象中由Lys46^1.46与Arg195^ECL2形成的离子锁,从而保持受体处于非活化状态。
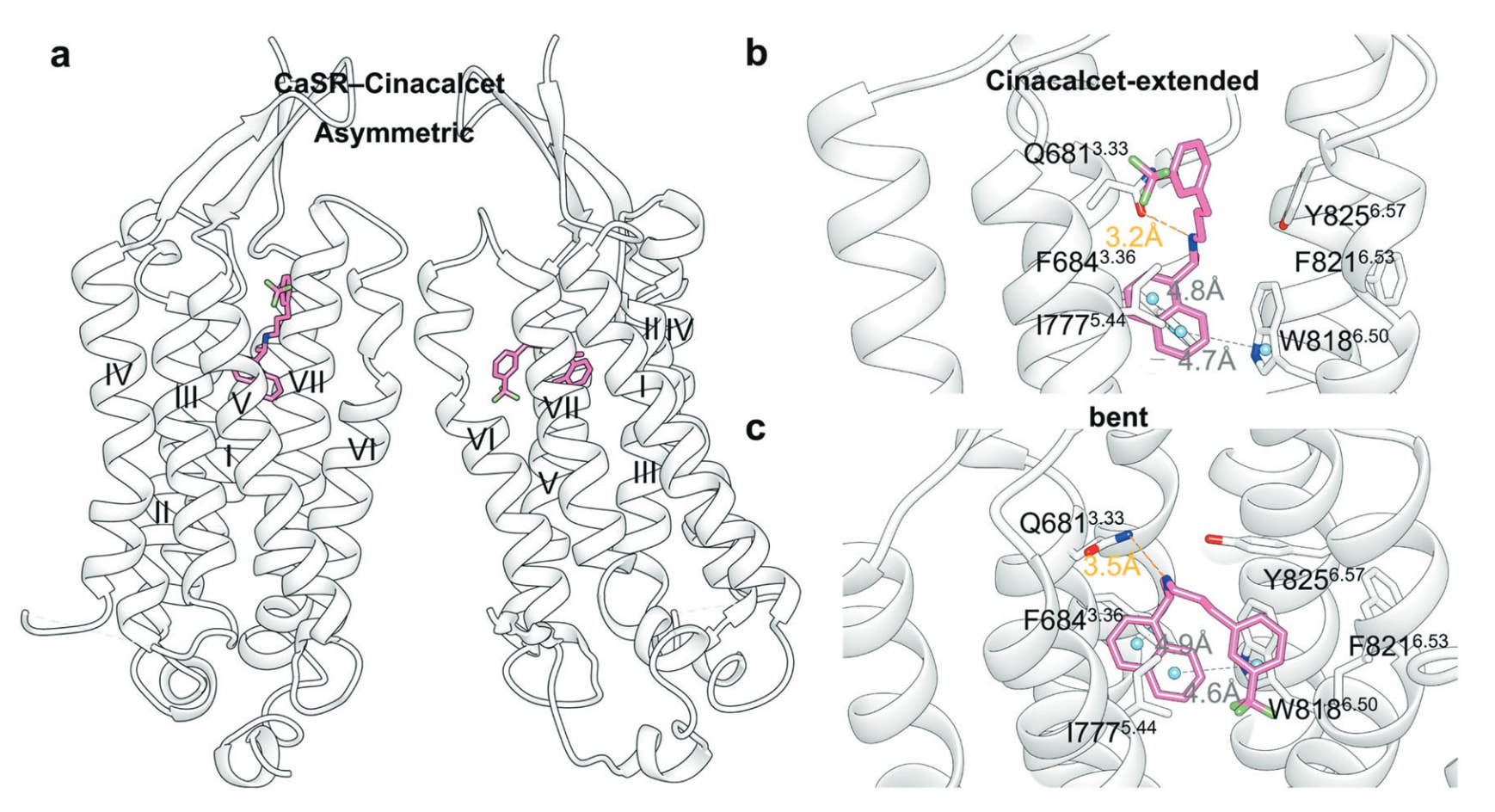
图18 a PAM cinacalcet结合CaSR的示意图(PDB: 7M3F)。b CaSR结合cinacalcet时呈“延展”构象的详细结合模式。c CaSR结合cinacalcet时呈“弯折”构象的详细结合模式,氢键以橙色虚线表示,π–π堆叠以灰色虚线表示。
- TM II–IV:CB1R−ORG27569、CB1R−ZCZ011 与 PAR2−AZ3451 结构
大麻素受体包括CB1R与CB2R两个亚型,由内源性大麻素激活,在突触传递的调控中发挥关键作用。其中CB1R是人脑中最丰富的GPCR,在多种生理功能中具有广泛分布和调控能力,因此被视为治疗多类中枢神经系统疾病的重要靶点。
ORG27569(39)(图20b)是首个被深入研究的CB1R负向变构调节剂(NAM)。尽管其能在体外调节CB1R信号,但在体内却难以有效调节正构大麻素的作用。CB1R−ORG27569与正构激动剂CP55940的三元晶体结构显示,ORG27569占据位于脂双层内侧、TM II与TM IV外侧的变构口袋(图22a)。其中,ORG27569的氯代吲哚环与Trp241^4.50形成关键芳香堆叠,并嵌入由His154^2.41与Val161^2.48构成的疏水口袋。Cys238^4.47、Trp241^4.50、Thr242^4.51与Ile245^4.54共同稳定其酰胺连接的哌啶苯基部分(图22b)。
近年来开发的ZCZ011(图20b)同样为吲哚类衍生物,表现出PAM及部分激动剂活性。在TM II–IV外侧口袋中,ZCZ011与受体形状高度互补(图22a)。其中,ZCZ011的吲哚环由Phe191^3.27锚定,与其主链形成氢键,并与侧链形成π–π堆叠(图22c)。此外,吲哚环还与来自TM2与TM4的Leu165^2.52、Ile169^2.56、Ile245^4.54与Val249^4.58形成疏水作用。
值得关注的是,NAM ORG27569、NAM AZ3451(38)以及PAM ZCZ011均结合在同一TM II–IV变构表面,但却产生相反的变构效应。ORG27569不同于传统NAM,它能增强激动剂亲和力,同时抑制Gi周转活性。
结构分析显示,ORG27569的抑制效应源于其稳定了由Phe155^2.42与Phe237^4.46构成的CB1R“激活开关”(图22d)。此外,ORG27569与Trp241^4.50形成的氢键,以及Trp241^4.50、Ser158^2.45与Ser206^3.42之间的氢键网络(图22b),共同维持受体处于失活构象。然而,其增强激动剂亲和力的具体机制尚未完全明确。
对比ZCZ011−CB1R结构与拮抗剂AM6538−CB1R结构发现,TM2中的Ile169^2.56向TM3发生明显偏移,使受体活性位点出现收缩(图22e),这一变化被认为与受体活化相关。此外,Ser173^2.60也向内移动并与CP55940形成氢键,从而进一步稳定正构激动剂的结合。
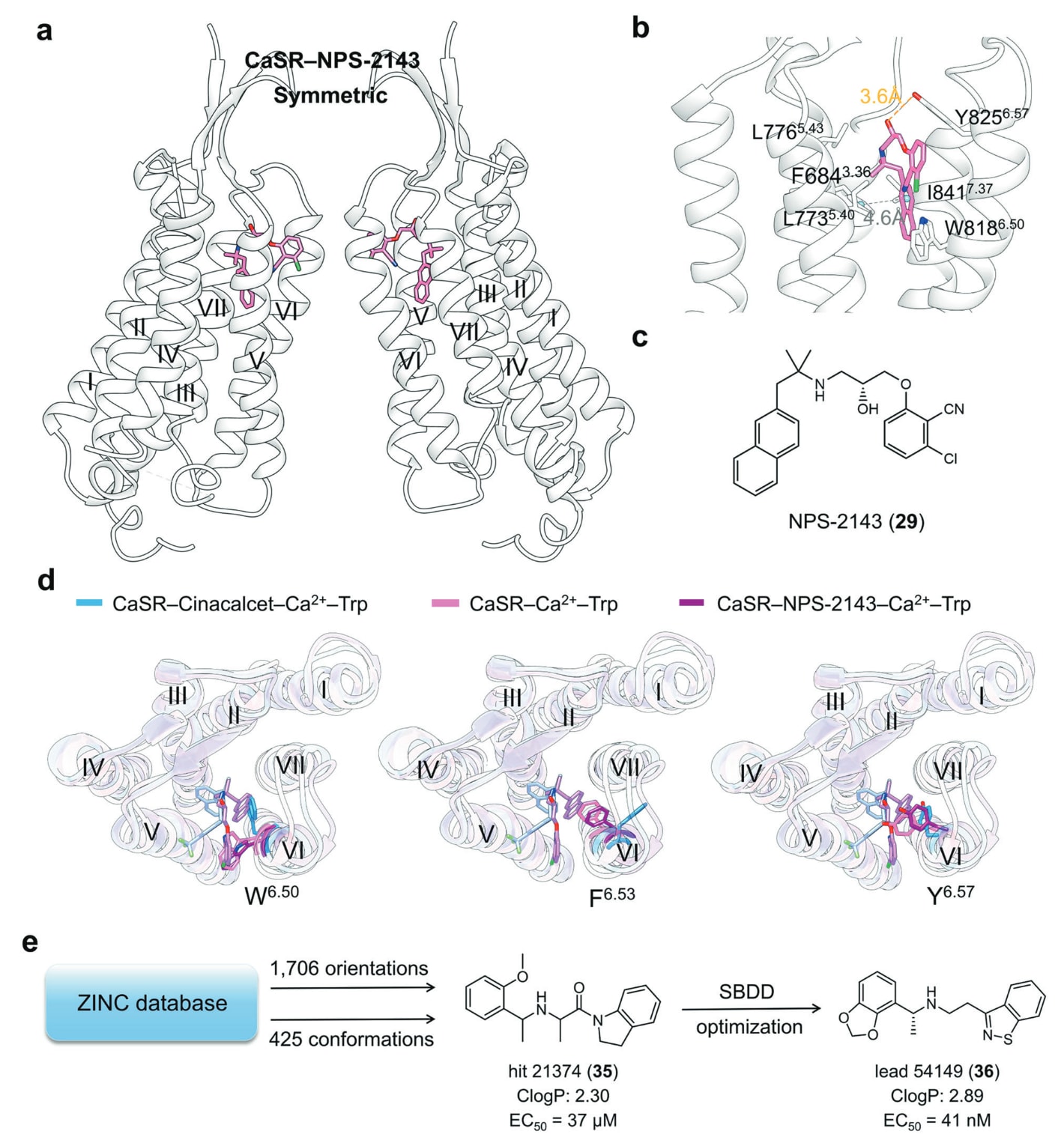
图19 a NAM NPS-2143 与 CaSR 结合的示意图(PDB: 7M3E)。b CaSR–NPS-2143 复合物的详细结合模式。氢键以橙色虚线表示,π–π 堆叠以灰色虚线表示。c 小分子变构配体 NPS-2143 的二维化学结构,用于清晰展示。d CaSR 在不同配体结合状态下关键残基的叠合结构:CaSR–Cinacalcet–Ca²⁺–L-Trp(蓝色卡通与蓝色棒,PDB: 7M3F);CaSR–Ca²⁺–L-Trp(粉色卡通与粉色棒,PDB: 7DD6);CaSR–NPS-2143–Ca²⁺–L-Trp(紫色卡通与紫色棒,PDB: 7M3E)用于展示 Trp818^6.50、Phe821^6.53、Tyr825^6.57 等关键残基在不同配体作用下的构象差异。e 利用 CaSR 结构信息发现新型 CaSR PAM 的流程示意,包括构象采样、虚拟筛选、片段拼接与立体化学优化等步骤。
表2|已解析的GPCR-合成变构调节剂复合结构:结合于7TMD外侧跨膜结构域的调节模式
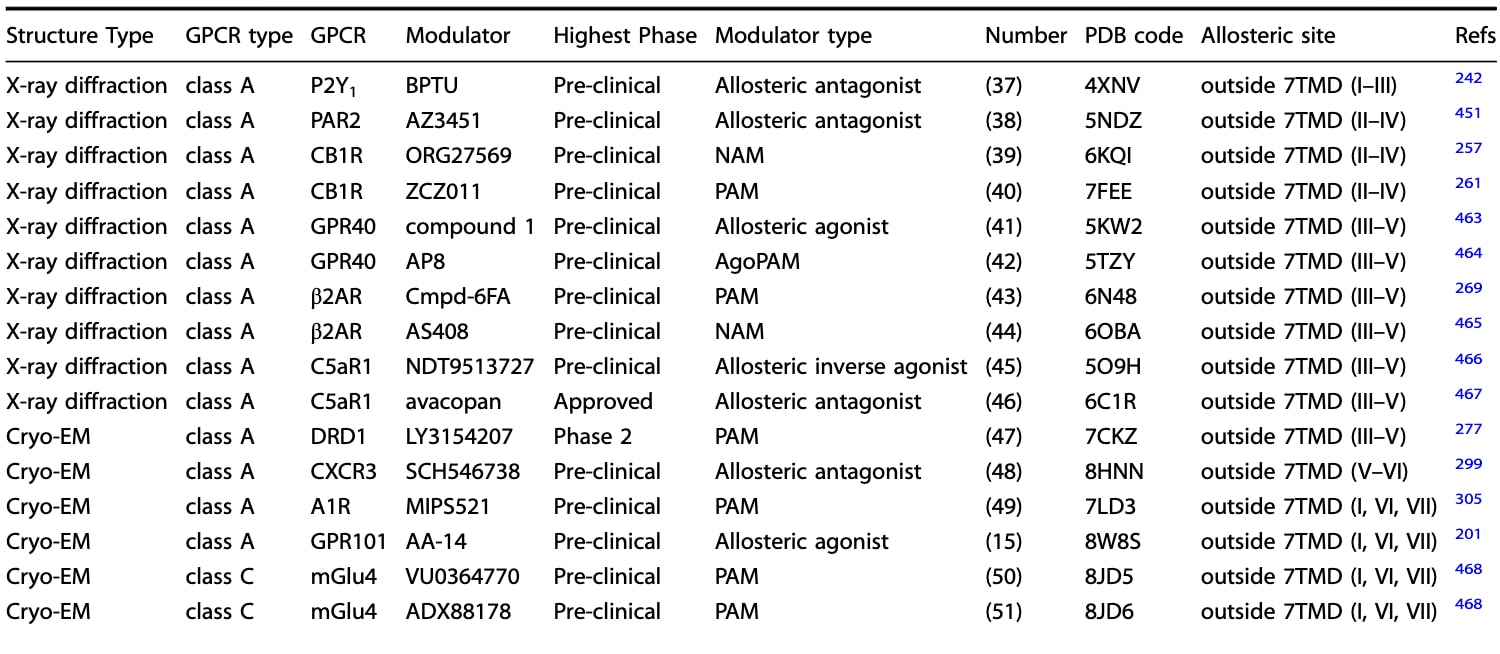
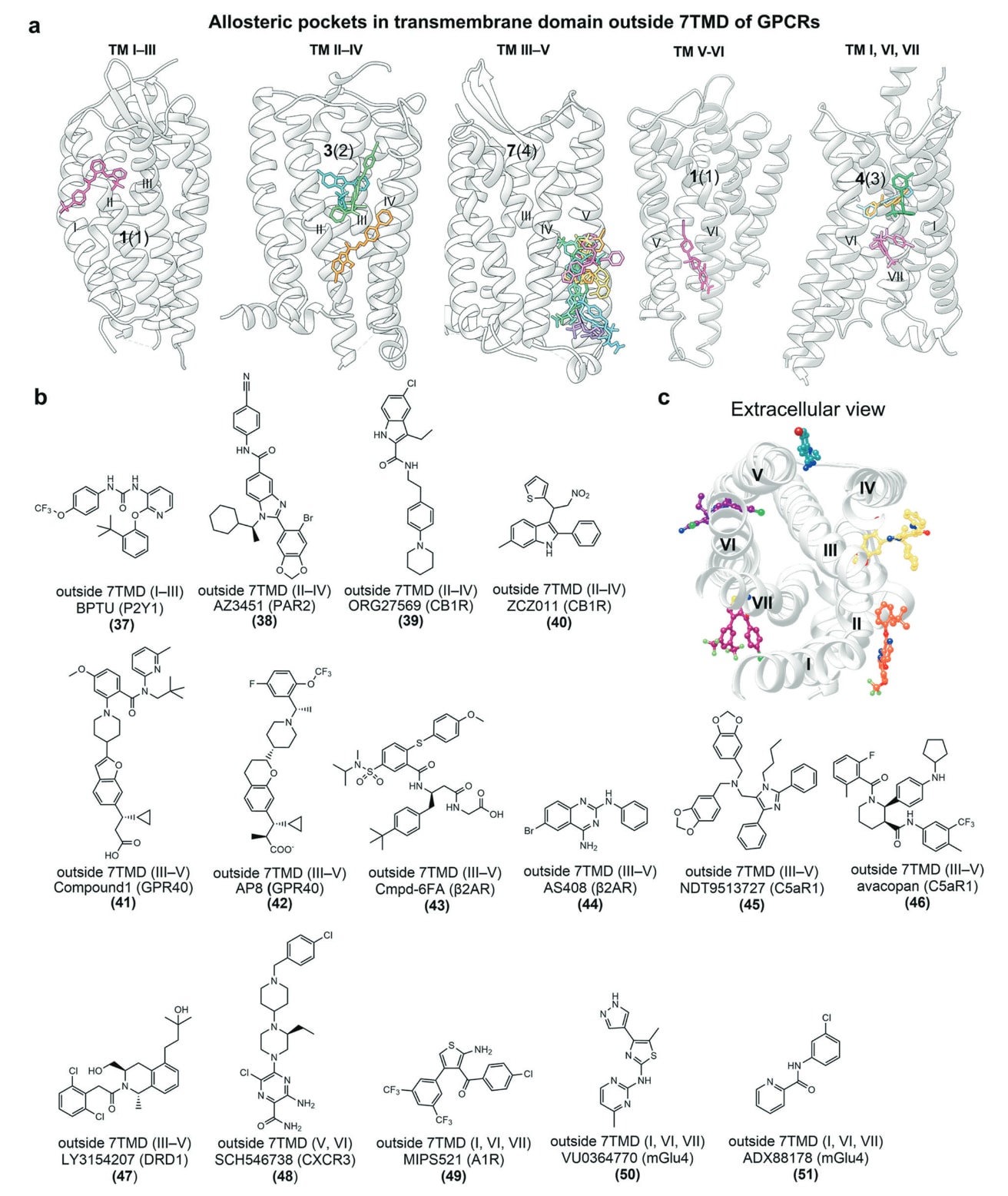
图20 a 展示了位于GPCR跨膜结构中、7TMD外侧的五种变构结合位点及其对应的小分子变构调节剂。图中以棒状模型呈现的小分子配体分别映射到五类代表性受体:位于7TMD外侧I–III区域的P2Y1(PDB: 4XNV)、外侧II–IV区域的CB1R(PDB: 6KQI)、外侧III与V区域的C5aR1(PDB: 6C1R)、外侧V与VI区域的CXCR3(PDB: 8HNN),以及外侧I、VI与VII区域的A1R(PDB: 7LD3)。每个结合口袋旁标注了其对应的独特调节剂数量(以粗体显示),并在括号中给出包含该口袋的GPCR数量。b 展示了这些靶向7TMD外侧跨膜结构的小分子合成变构配体的二维化学结构。c 为上述五个变构结合位点的胞外侧视图。
- TM III–V:GPR40–compound 1、GPR40–AP8、β2AR–AS408、β2AR–Cmpd-6FA、C5aR1–NDT9513727、C5aR1–avacopan以及DRD1–LY3154207结构
在多种GPCR中,跨膜螺旋TM III–V外侧普遍存在一个深口袋,可容纳一类结合于此的变构调节剂。在这一口袋中,变构激动剂与PAM(如compound 1、AP8、Cmpd-6FA与LY3154207)会定位在靠近ICL2的区域,并通过直接作用稳定ICL2的α螺旋,促进ProICL2向内运动。这会引发TM III约3°的向内偏移,继而带动TM V与TM VI向外移动,这是GPCR活化的典型结构特征。因此,变构调节剂的结合能够提升受体处于活化构象的比例,从而提高其对激动剂的亲和力。相反,变构拮抗剂与NAM(如AS408、NDT9513727与avacopan)远离ICL2结合,由于其靠近TM V的脯氨酸折角,受体–配体相互作用会共同抑制TM III、TM IV与TM V之间必需的相对运动,阻碍受体活化。
多巴胺D1受体是典型案例。多巴胺是关键的儿茶酚胺类神经递质,通过D1–D5受体发挥信号功能,其中D1受体调控中枢神经系统中的神经元生长、学习与记忆。LY3154207是D1受体的选择性PAM,并在2022年的Ⅱ期临床试验中改善了路易体痴呆相关的运动症状。研究揭示LY3154207与D1受体存在两种结合模式,分别呈现upright与boat构象。在这两种结构中,LY3154207均位于受体与脂质双层界面,由TM III、TM IV与ICL2包围。upright构象下,其叔醇基团朝向TM III并与Cys1153.44形成单一氢键,同时其四氢异喹啉环与Val1193.48、Trp1233.52、Leu1434.45形成疏水作用,二氯苯基部分与Arg130ICL2形成π-阳离子作用。boat构象下,二氯苯基既与Arg130ICL2形成π-阳离子作用,又与Trp1233.52形成夹层式π-π堆积;四氢异喹啉环则与Met135ICL2、Ala1394.41、Ile1424.44、Leu1434.45及Lys134ICL2的烷基链形成疏水作用,并与Arg130ICL2、Lys1384.40形成两条氢键。结构叠合显示,多巴胺的结合模式基本不变,而LY3154207通过作用于Arg130ICL2与Lys134ICL2稳定ICL2,从而提升D1受体活化构象的比例。
另一个结合于TM III–V外侧的变构调节剂是C5aR1的变构拮抗剂avacopan。C5aR1是结合炎症介质C5a的关键受体,主要分布于多类免疫细胞表面。C5aR1–C5a通路的过度活化会引发失控炎症,因此C5aR1拮抗剂被视为多种炎症性疾病(包括脓毒症与COVID-19)的潜在治疗手段。Avacopan于2021年被FDA批准用于治疗重症ANCA相关性血管炎,并具有偏向性抑制β-arrestin耦联的能力。C5aR1–avacopan复合物结构显示其结合位点位于TM III与TM V之间的7TMD外侧。Avacopan的环戊烷基深入TM III与TM IV之间的缝隙,与Leu1253.41、Val1594.48、Leu1634.52与Leu1674.56构成疏水作用。邻甲基三氟甲基苯基在TM III与TM V之间与Ile1243.40、Leu1253.41、Leu2095.45、Trp2135.49、Pro2145.50与Leu2185.45形成疏水与芳香作用;间甲基氟苯基更深层结合,并与Phe1353.52、Ile2205.56、Cys2215.57与Phe2245.60形成非极性作用。仅有一条氢键来自其酰胺羰基与Trp2135.49,另有一条经水介导的极性作用连接至Thr2175.53。在非活化状态的C5aR1中,Trp2135.49需要通过构象变化来容纳avacopan。Avacopan可能通过疏水作用稳定Ile1243.40、Pro2145.50与Phe2516.44的非活化构象,从而阻碍TM5与TM6的活化所需运动。
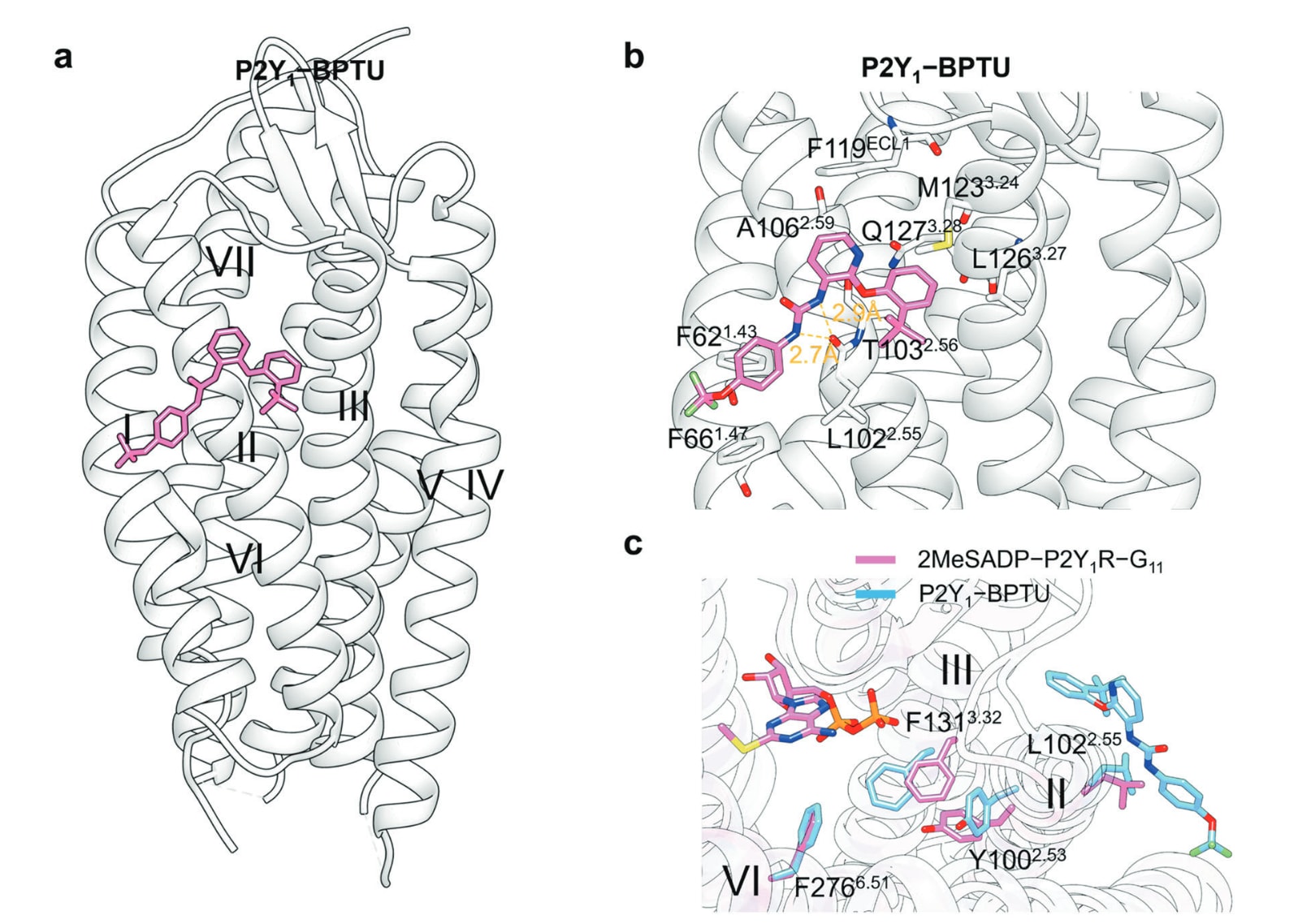
图21 a 展示了变构拮抗剂BPTU与P2Y1受体结合的示意图(PDB: 4XNV)。b 描述了BPTU与P2Y1受体的具体结合模式,其中氢键以橙色虚线表示。c 对比叠合了2MeSADP−P2Y1R−G11结构(粉色示意)与P2Y1R−BPTU结构(蓝色示意)中被重点标出的残基,展示两种配体结合下受体局部构象的差异(PDB: 7XXH与4XNV)。
- TM V–VI:CXCR3–SCH546738结构
C–X–C趋化因子受体3(CXCR3)属于A类GPCR,在效应T细胞上高水平表达,并可被CXCL9、CXCL10与CXCL11激活。由于其在Ⅰ型免疫中的关键作用,研究中已合成多种靶向CXCR3的激动剂与拮抗剂,用于感染、自身免疫疾病、移植排斥及肿瘤等的潜在治疗。在这些分子中,SCH546738表现尤为突出,其通过高效抑制T细胞趋化激活,在多项临床前研究中展现了显著效果,亲和力达到0.4 nM。
在CXCR3–SCH546738复合物结构中,SCH546738被固定在由TM3、TM5与TM6围成的狭窄疏水口袋内。其头部被Phe1353.36、Ala1393.40、Phe2245.47、Leu2285.51、Met2315.54、Ile2616.41与Ala2736.53构成的疏水环境包围;尾部则向脂质双层方向延伸,与Tyr2355.58、Leu2395.62与Leu2586.38发生作用。由于其结合位点的独特性,SCH546738的嵌入可能削弱TM5与TM6之间的紧密堆积,从而使受体维持在非活化状态。
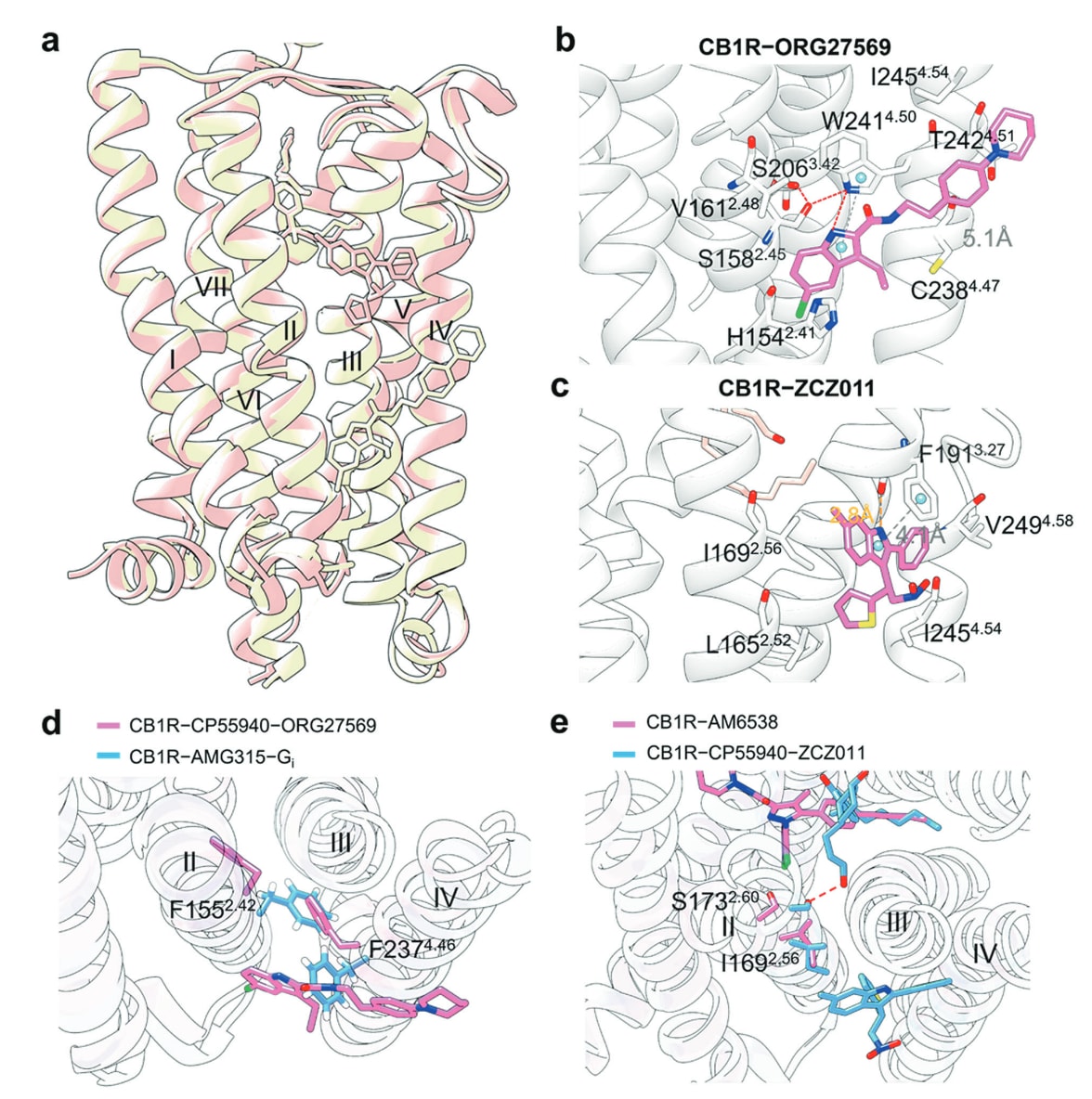
图22 a 展示了NAM ORG27569(黄色,PDB: 6KQI)与PAM ZCZ011(粉色,PDB: 7FEE)在正构配体结合的CB1R上的共晶结构叠合图,用以对比两类变构分子在受体中的结合位置与构象差异。b 描述了ORG27569与CB1R的详细结合模式,包括其在口袋中的定位与关键相互作用。c 展示了ZCZ011与CB1R的结合方式,其中氢键以橙色虚线标示,π–π堆积以灰色虚线呈现。d 叠合显示了CB1R–CP55940–ORG27569结构(粉色)与CB1R–AMG315–Gi结构(蓝色)中被重点标出的残基差异,体现变构结合对受体构象的影响(PDB: 6KQI与8GHV)。e 对比叠合了CB1R–AM6538结构(粉色,PDB: 5TGZ)与CB1R–CP55940–ZCZ011结构(蓝色,PDB: 7FEE)中关键残基的位置变化,呈现ZCZ011结合对正构口袋及邻近区域构象的调控作用。
- TM I、VI、VII:A1R–MIPS521、GPR101–AA-14、mGlu4–VU0364770与mGlu4–ADX88178结构
腺苷A1受体(A1R)是腺苷受体的一个亚型,一直被视为开发非阿片类镇痛药的重要靶点,用于治疗慢性疼痛。然而,目前尚无选择性且已获临床批准的A1R激动剂或拮抗剂。MIPS521是一种A1R的PAM,能够抑制脊髓伤害性感觉信号,并在大鼠模型中展现镇痛效果,其pEC50为6.9±0.4。结构研究显示,MIPS521结合于由TM I、TM VI与TM VII围成的、位于7TMD外侧的变构口袋中。该口袋由Leu181.41、Ile191.42、Val221.45、Leu2426.43、Leu2456.46、Ser2466.47、Phe2757.40、Leu2767.41与Met2837.48等残基形成浅层疏水腔。MIPS521的氨基与Ser2466.47与Leu2767.41主链羰基形成氢键。对比ADO–A1R–Gi2与MIPS521–ADO–A1R–Gi2结构显示,MIPS521几乎不影响ADO的结合位置,也未显著改变受体整体构象。其作用机制可能源于其与变构口袋的结合可稳定A1R的活化构象,并进一步促进邻近、保守且与A类GPCR活化相关的Na+口袋的塌陷。
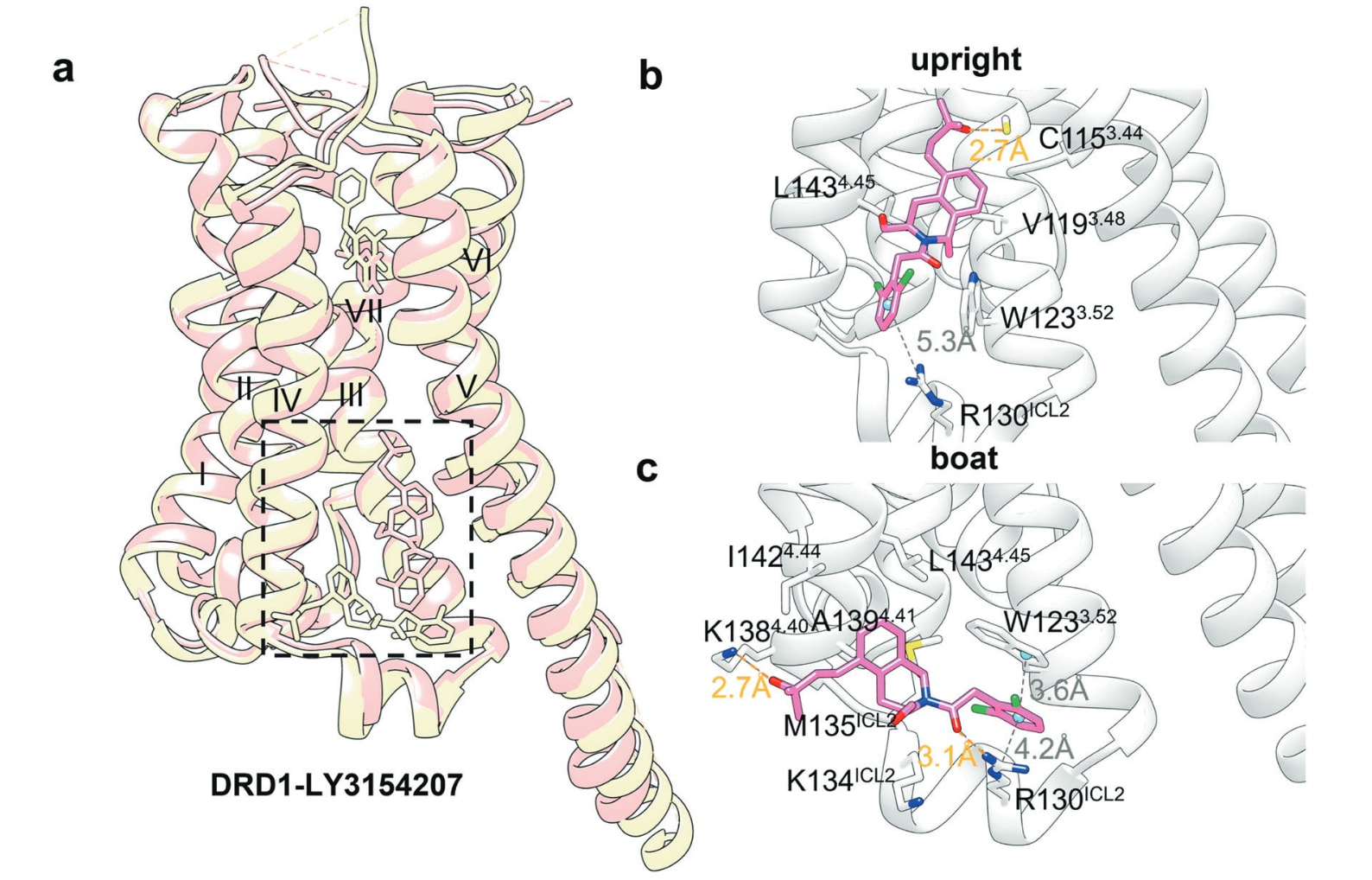
图23 a 展示了PAM LY3154207与多巴胺D1受体复合物在upright构象(粉色,PDB: 7CKZ)与boat构象(黄色,PDB: 7LJC与7X2F)中的共晶结构叠合图,用以对比其两种结合姿态的空间差异。b 描述了LY3154207在upright构象下与D1受体的详细结合模式,包括关键疏水作用、氢键及芳香相互作用,其中氢键以橙色虚线标示。c 展示了LY3154207在boat构象下的结合模式,重点呈现其与受体残基之间的π–π堆积及π–阳离子作用(以灰色虚线表示),以及与周围残基形成的其他相互作用。

图24 a 展示了变构拮抗剂avacopan与C5a受体1结合的示意图(PDB: 6C1R)。b 描述了avacopan与C5a受体1的具体结合模式,其中氢键以橙色虚线标示。c 叠合呈现了在C5aR1–PMX53–avacopan结构(粉色,PDB: 6C1R)与C5aR1–C5a–Go结构(蓝色,PDB: 8IA2)中,关键残基Trp2135.49的构象差异。
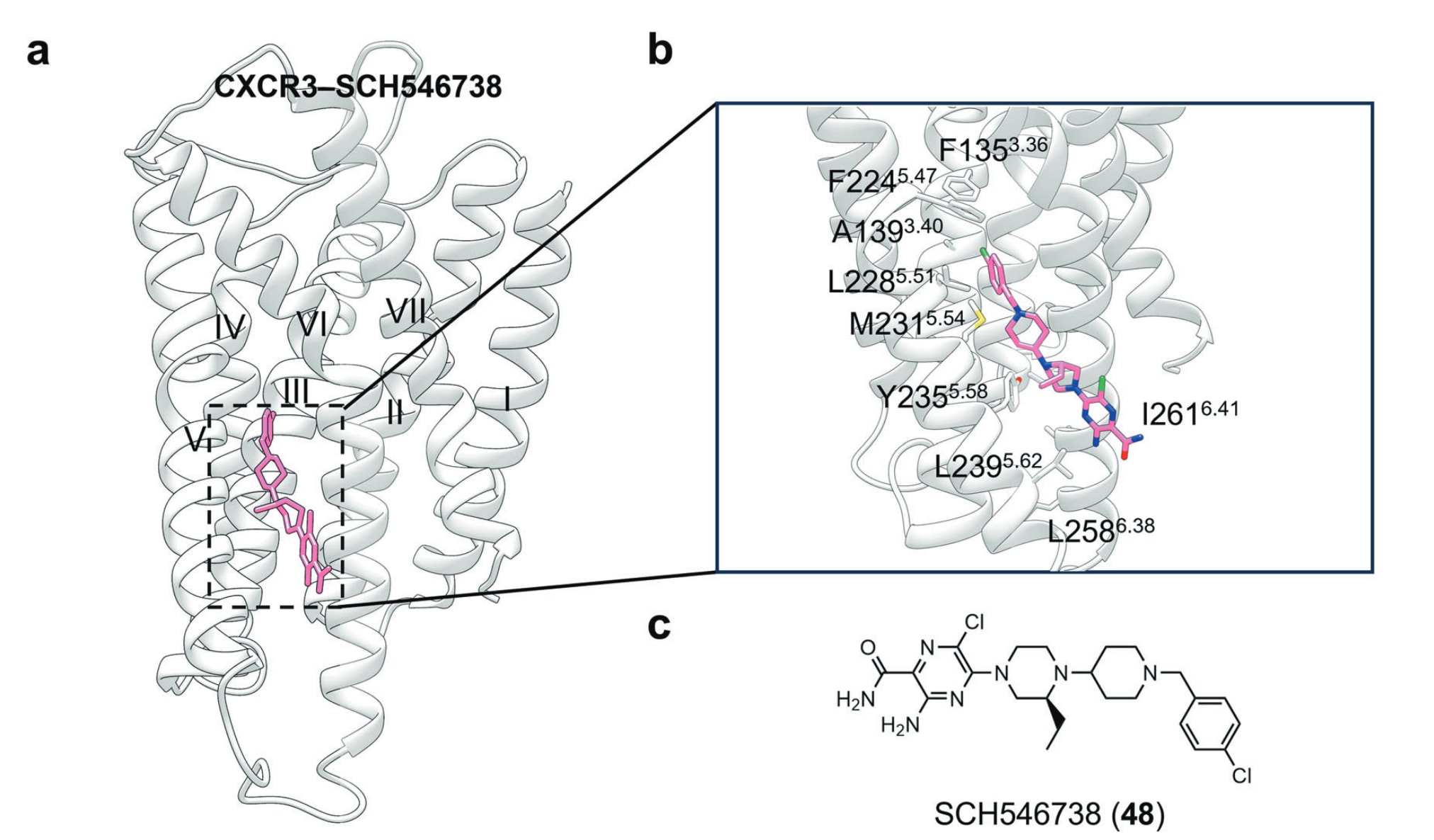
图25 a 展示了变构拮抗剂SCH546738与CXCR3结合的示意图(PDB: 8HNN)。b 描述了SCH546738在CXCR3中的详细结合模式,包括其在疏水口袋中的定位与关键相互作用。c 为SCH546738的小分子二维结构,以辅助理解其化学骨架与结合构象。
5.3.5 靶向GPCR跨膜结构域(7TMD内部)
- 7TMD内部:FFAR3–AR420626、CRF1R–CP-376395、mGluR5–mavoglurant、mGluR5–compound 14、mGluR5–HTL14242、mGluR5–Fenobam、mGluR5–M-MPEP与PTH1R–PCO371结构
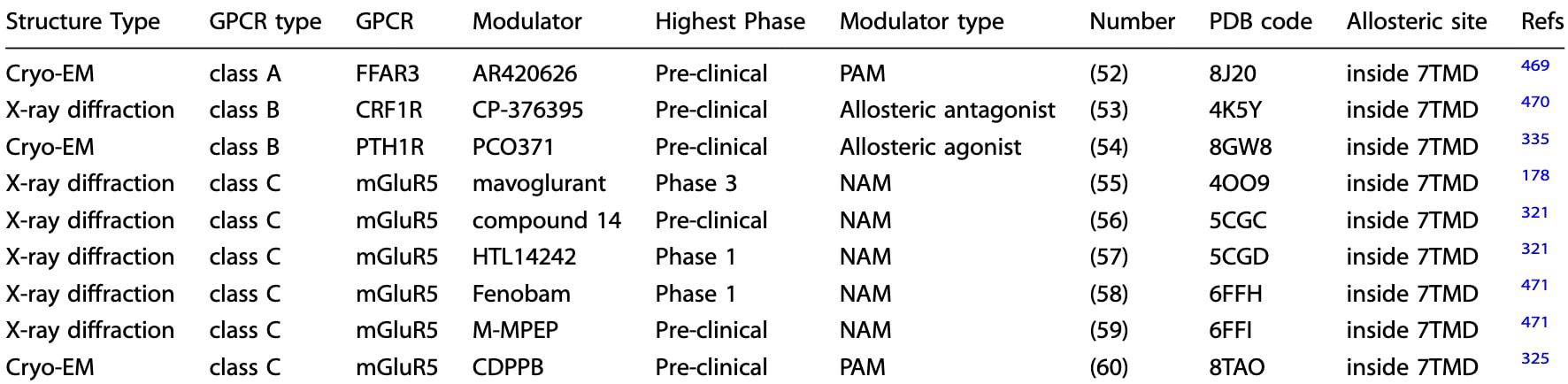
代谢型谷氨酸受体5(mGluR5)是八种mGlu受体中在脑内表达最广的亚型之一。近年来,其被证实参与多种认知与精神疾病,包括精神分裂症。mGlu受体家族在C类GPCR中在变构药理方面尤为丰富,其中mGlu5受体的变构调节更因其在帕金森病与精神分裂症等疾病中的潜在治疗价值而备受关注。mGlu5的NAM可在不阻断生理性谷氨酸功能的前提下抑制过度活化的信号,因此被视为多类神经系统疾病的候选治疗分子。其关键结合决定因素位于TM III、TM VI与TM VII。HTL14242是其中进展最快的口服NAM之一,正处于肌萎缩侧索硬化症的早期临床研究中。mGluR5与HTL14242先导化合物compound 14的晶体结构揭示了SAR优化的依据:其嘧啶连接基贯穿由Tyr6593.44、Ser8097.39、Val8067.36与Pro6553.40组成的变构口袋狭窄通道,苯环与吡唑环分别填入两个亚口袋。其中,苯环上的腈基至关重要,其空间指向恰好允许通过一枚水分子与Val7405.40形成氢键。因此移除或替换腈基会显著降低亲和力。吡唑端的构象则可容纳略大环结构与少量取代,只要保持氮原子与Ser8097.39的氢键以及周围极性网络即可。基于此类SBDD研究,HTL14242被成功设计出来,在保持与compound 14相似结合方式的同时,具备更佳的理化与药代性质。进一步比较mGlu5-NAM结构后发现,NAM普遍增强TM III与TM VI胞质端的相互作用,形成Lys6653.50−Glu7706.35的盐桥,从而阻碍TM VI向外运动。而在PAM复合物中则不见此相互作用。此外,保守残基Trp7856.50会发生构象变化以调整变构口袋大小并适应配体结合。该残基在A类GPCR中作为“开关”发挥作用,可能在C类GPCR中也具有类似功能,但仍需进一步研究。
结合于跨膜结构域的另一类变构调节剂为PTH1R的变构激动剂。甲状旁腺激素受体PTH1R属于B1类GPCR,由PTH与PTH相关多肽激活,是维持矿物质离子稳态与骨代谢的核心因子。近年来,一种高度选择性的、口服小分子PTH1R激动剂PCO371被发现,目前正在用于治疗甲旁腺功能减退症的Ⅰ期临床试验中。PCO371由四部分构成:三氟甲氧基苯基、螺环咪唑啉酮、二甲基苯基与二甲基海因。PTH1R–PCO371复合物结构显示其结合口袋由TM II、TM III、TM VI与TM VII包围,与mGluR5类似。PCO371中螺环咪唑啉酮的NH与Tyr4597.57形成氢键;二甲基海因的羰基则与Arg2192.46的质子化氮形成盐桥。此外,其三氟甲氧基苯基、螺环咪唑啉酮、二甲基苯基与二甲基海因分别与多处疏水残基作用。值得注意的是,Gs蛋白中的Glu394G.H5.24与其羰基形成氢键,而Tyr393G.H5.23与其形成疏水作用,从而稳定PCO371–PTH1R–Gs三元复合物。由于PCO371结合后会阻止内源性配体进入7TM核心,因此目前仅有PCO371单独结合PTH1R的结构。PCO371的结合会使TM6胞外与胞质端向内移动。对比PCO371–PTH1R与CP-376395–CRF1R结构可见,PCO371结合位点更深,因此不会像CP-376395那样阻碍构象保守的Pro6.47−X−X−Gly6.50折角,也不会干扰结合于细胞内侧的G蛋白或arrestin。目前,PCO371可能是唯一能通过直接与下游转导蛋白作用来稳定受体活化状态的变构激动剂。
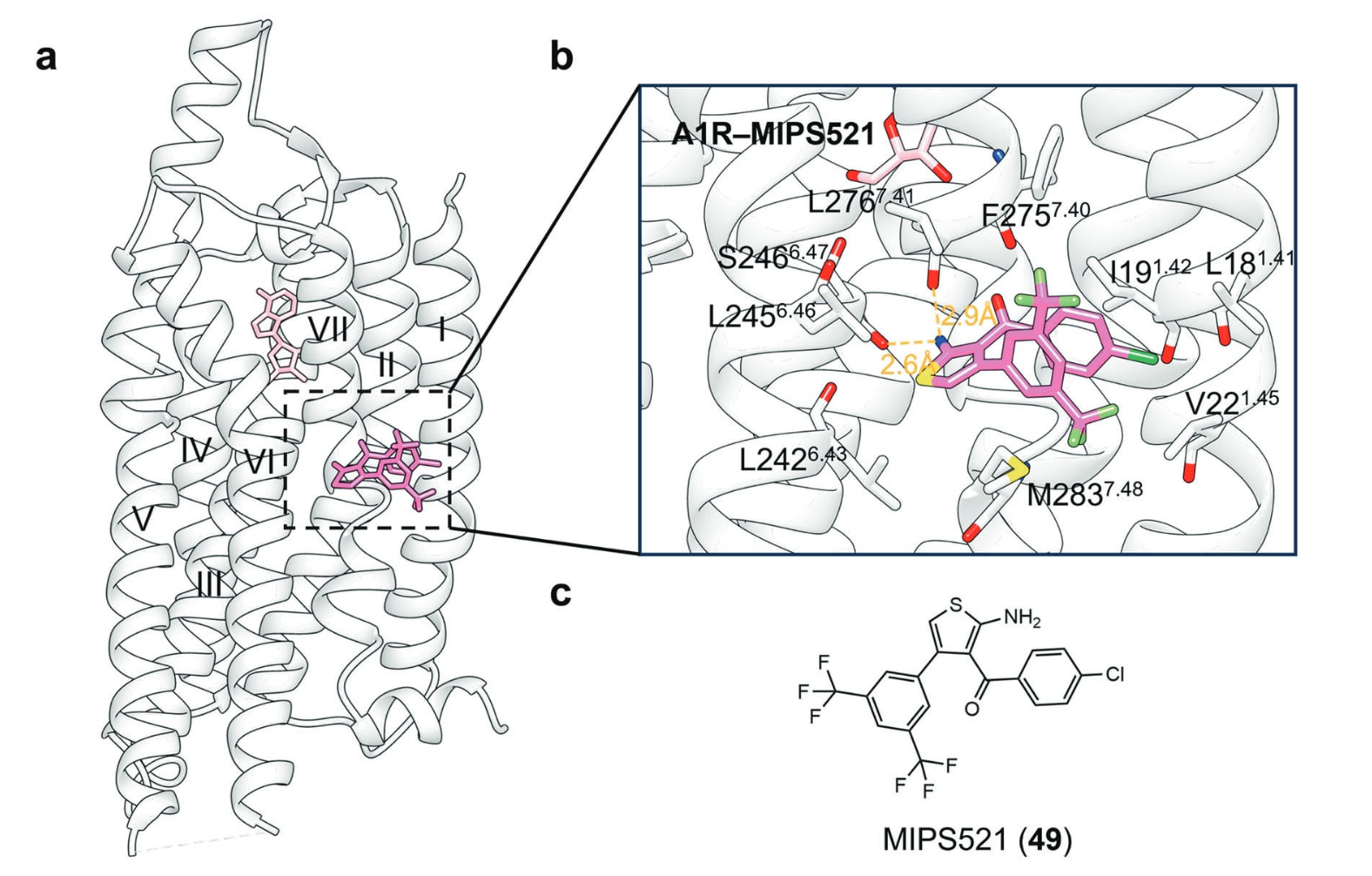
图26 a 展示了PAM MIPS521与A1R结合的示意图(PDB: 7LD3)。b 描述了MIPS521在A1R中的具体结合方式,其中氢键以橙色虚线标示。c 展示了小分子变构配体MIPS521的二维化学结构,以便更清晰理解其构成。
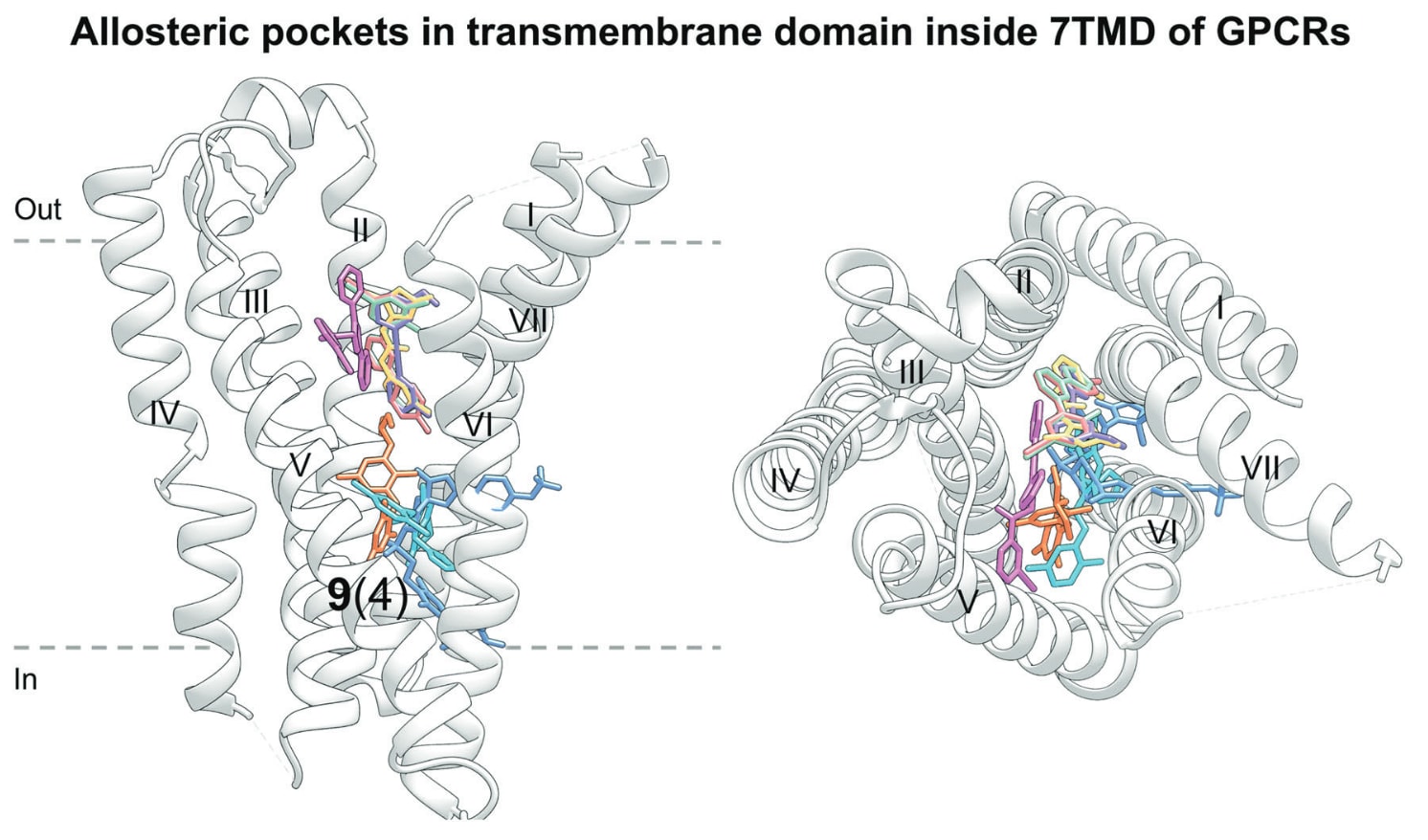
图27 展示了GPCR跨膜结构域中、位于7TMD内部的变构结合位点及其对应的小分子变构调节剂。图中以棒状模型显示小分子配体,并映射至代表性受体CRF1R(PDB: 4K5Y)。每个结合口袋旁标注了独特调节剂数量(粗体)以及包含该口袋的GPCR数量(括号中)。
图28 展示了靶向GPCR 7TMD内部跨膜结构域的各类小分子合成变构配体的二维化学结构。
表3|与合成变构调节剂在7TMD内部跨膜结构域结合的GPCR复合物结构已被解析
5.3.6 靶向GPCR胞内表面(7TMD外侧与内侧)
在这些情况下,变构调节剂会结合于7TM螺旋束胞质端与下游信号转导蛋白之间的界面区域,包括7TMD的外侧与内侧。迄今为止,依据已解析的晶体结构,仅在GPCR胞内表面发现了两个不同的结合位点(见Table 4),分别为位于TM V−VII外侧的口袋,以及位于7TMD内部的口袋(Fig. 31)。
- 7TMD外侧(V−VII区域):GPR88−2-PCCA、GCGR−MK-0893、GCGR−NNC0640、GLP-1R−PF-06372222、GLP-1R−compound 2、GABAB−GS39783与GABAB−BHFF结构
已报道的、位于7TMD外侧胞内表面的变构结合位点均附着于TM VI,并位于TM V与TM VII之间,提示TM VI在将配体结合信息传递至正构口袋中具有功能性作用。G蛋白耦联需要TM VI胞内端向外运动,这是GPCR活化的关键结构特征,因此该区域成为设计小分子变构调节剂的策略性靶点。
GABAB受体属于C类GPCR,以GB1与GB2组成的异源二聚体形式存在,可被中枢神经系统主要抑制性递质GABA激活,是抑郁、精神分裂症与药物成瘾等多类神经疾病的重要治疗靶点。二聚体的形成带来一种特有的二聚体变构口袋,使GS39783与BHFF无法作用于单体,而只能调控二聚体活性。BHFF是一种有效的PAM,仅在正构激动剂存在时提高GABAB活性,但在小鼠中未表现出显著效力。其与GABAB受体的复合物结构显示,BHFF位于GB1的TM V–TM VI与GB2的TM VI胞内端之间。BHFF的3-羟基与酮基分别与GB1的Lys792ICL3侧链形成两条氢键,同时整体埋藏于由GB1与GB2多处疏水残基构成的腔体中。与另一种PAM GS39783相比,两者结合位置高度相似,引发的整体构象几乎一致。相较于仅结合正构激动剂,PAM会促使GB2胞内端TM3与TM5变直并向内移动,同时稳定TM6介导的二聚化,从而促进受体活化。
Compound 2是GLP-1R的一种新型covalent ago-PAM,可与TM VI上的Cys3476.36形成共价键,表现出部分甚至接近完全的激动活性,但因药代性质欠佳而在临床试验中失败。在其共晶结构中,compound 2位于TM VI的膜侧表面,并仅与TM VI上的残基发生作用,其中其磺酰基与Cys3476.36形成二硫键。其叔丁基部分朝向TM VII并与Ala3506.39、Lys3516.40形成疏水作用,而二氯喹喔啉部分朝向ICL3并与Lys3466.35、Cys3476.36形成范德华作用。仅compound 2、仅GLP-1以及二者共同结合均能促使TM VI胞质端外移,而在compound 2–GLP-1–GLP-1R–Gs复合结构中,额外出现了两组长距离盐桥,有助于增强G蛋白结合。此外,GLP-1R的N端α螺旋因compound 2的结合而向下插入正构口袋,进一步稳定活化构象。共价结合提高了分子解离的能垒,从而减少潜在的脱靶作用,提示跨膜螺旋中突出的游离半胱氨酸可能是构建不可逆GPCR变构调节剂的机会。
所有已报道的变构拮抗剂与NAM(MK-0893、NNC0640、PF-06372222)均位于TM VI靠近TM VII的另一侧,其结合位点及模式高度保守。此处TM VI突出的结构可作为卡钳式调节剂的依托,需具备足够的跨螺旋裂缝以容纳配体,并依赖稳固的疏水作用保持结合。其结合会限制TM VI向外运动,从而阻止受体活化。
胰高血糖素受体(GCGR)属于B类GPCR,在维持葡萄糖稳态中发挥关键作用。鉴于胰高血糖素升高血糖的功能,GCGR小分子拮抗剂被视为糖尿病治疗的潜在策略。MK-0893作为GCGR的变构拮抗剂已进入Ⅱ期临床,但因增加LDL-C等副作用而受限。其与GCGR的X射线结构显示,MK-0893位于TM V–VII外侧的胞内口袋中。其末端羧酸与Arg3466.37形成盐桥,并参与与Asn4047.61、Lys4057.62的氢键网络,以及与Ser3506.44的水介导氢键。其酰胺基分别与Ser3506.41、Leu3997.56及Lys3496.40形成氢键,而甲氧基萘基嵌入TM V与TM VI间的腔体,形成多处疏水作用与π–阳离子作用。该结构与其他非活化态GCGR高度一致,未表现出由MK-0893引发的特异构象。
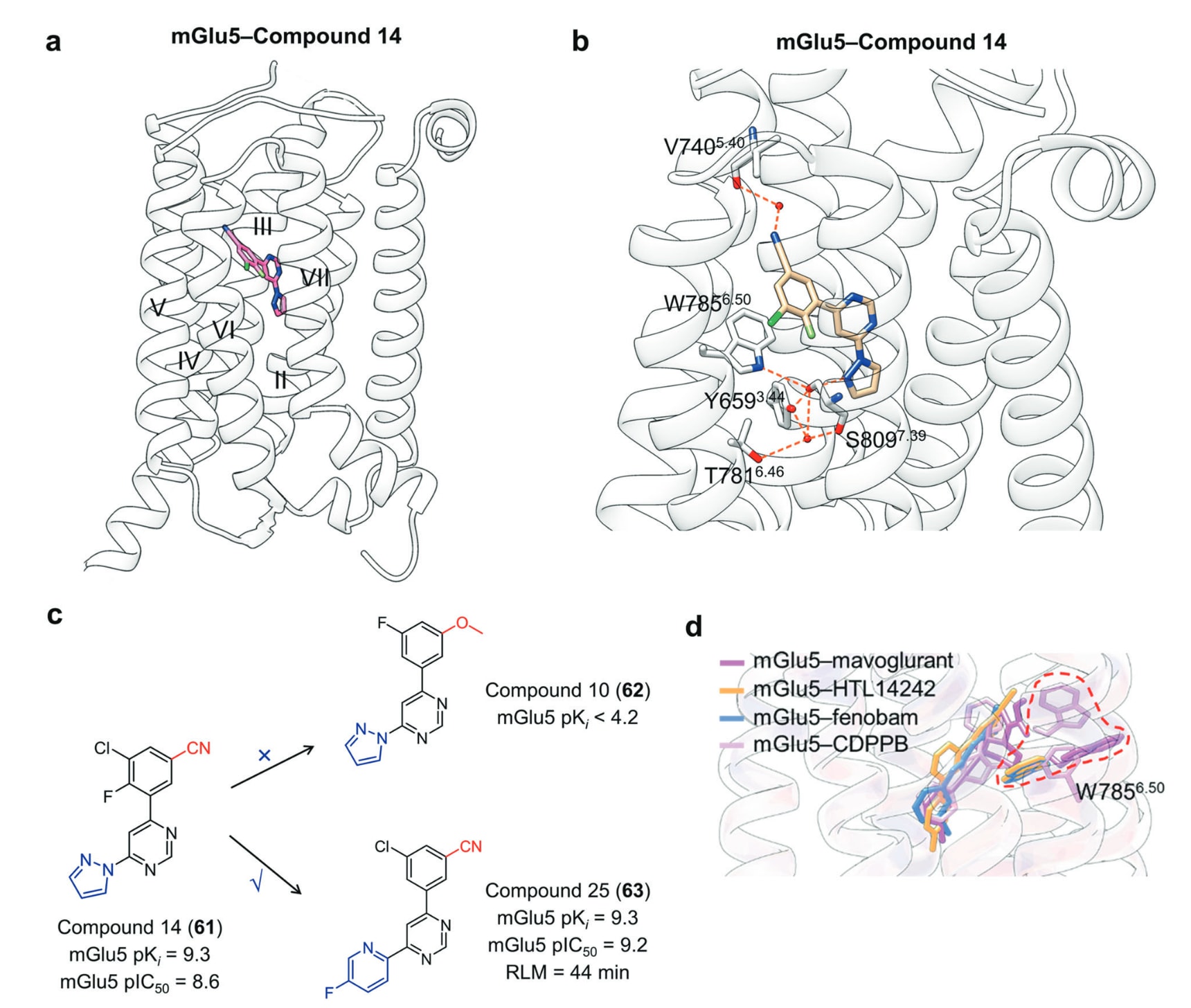
图29 a 展示了NAM fenobam与mGlu5受体结合的示意图(PDB: 5CGC)。b 描述了mGlu5受体与compound 14的详细结合模式,图中以橙色虚线标示极性相互作用。c 展示了mGlu5受体NAM的构效关系(SAR)优化过程。d 叠合展示了mGlu5–mavoglurant(紫色,PDB: 4OO9)、mGlu5–HTL14242(橙色,PDB: 5CGD)、mGlu5–fenobam(蓝色,PDB: 6FFH)以及mGlu5–CDPPB(粉色,PDB: 8TAO)结构中被重点标出的残基,用于对比不同变构配体结合下受体局部构象的差异。
图30 a 展示了变构激动剂PCO371与PTH1R结合的示意图(PDB: 8GW8)。b 描述了PCO371与PTH1R的具体结合模式,其中氢键以橙色虚线表示。c 叠合显示了结合PCO371的PTH1R(粉色)与结合PTH的PTH1R(蓝色)之间的构象差异,呈现PCO371结合后受体结构发生的变化。
表4|展示了已解析的、与合成变构调节剂结合于胞内表面的GPCR复合结构列表。

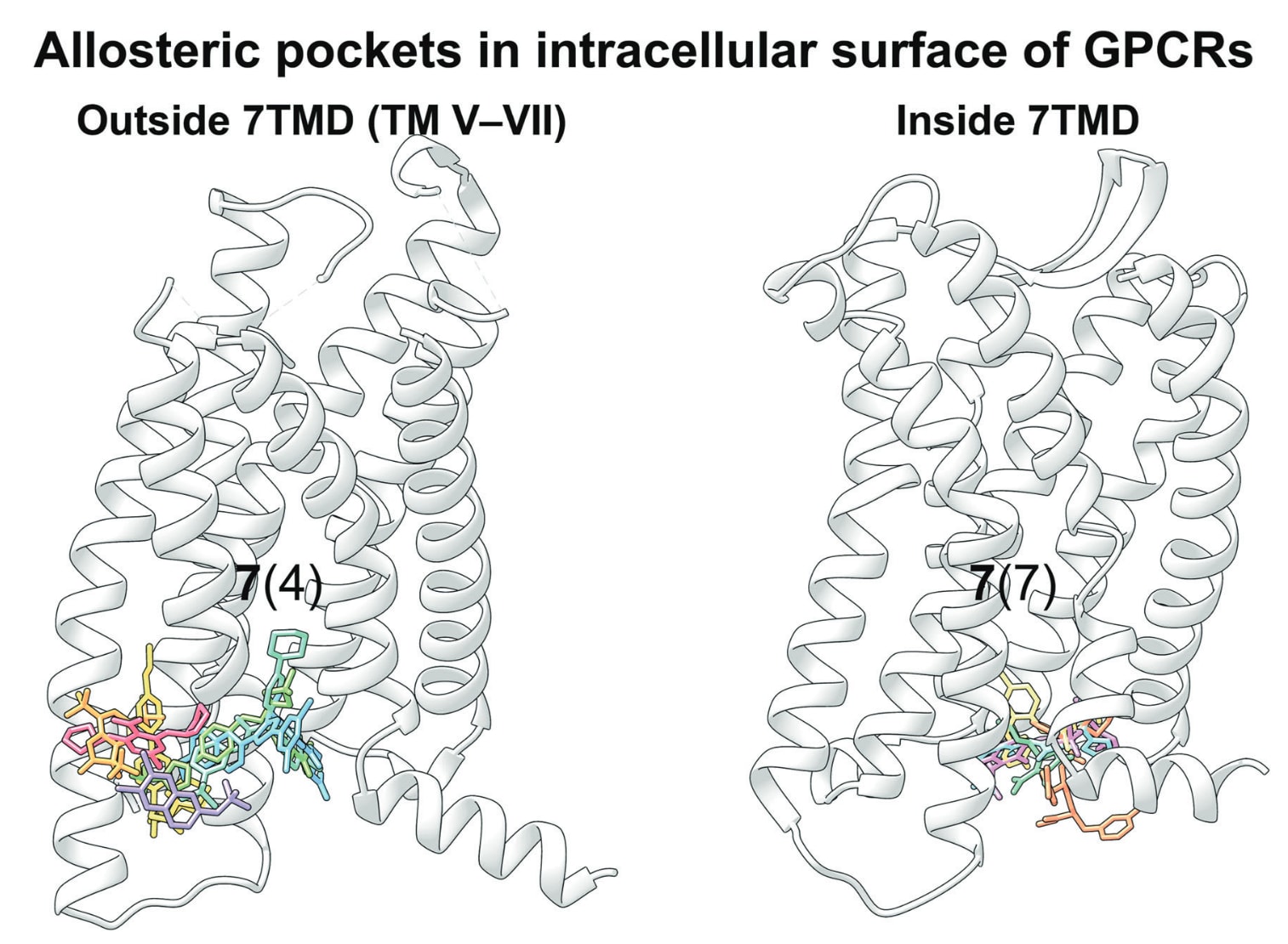
图31|展示了GPCR中两类胞内变构结合位点及其对应的小分子变构调节剂。 图中以棒状模型呈现的小分子配体分别映射到代表性受体:位于7TMD外侧(V−VII区)的GCGR(PDB: 5EE7)以及位于7TMD内部的CCR2(PDB: 5T1A)。每个结合口袋旁均标注了独特调节剂的数量(以粗体显示)以及包含该口袋的GPCR数量(括号中)。
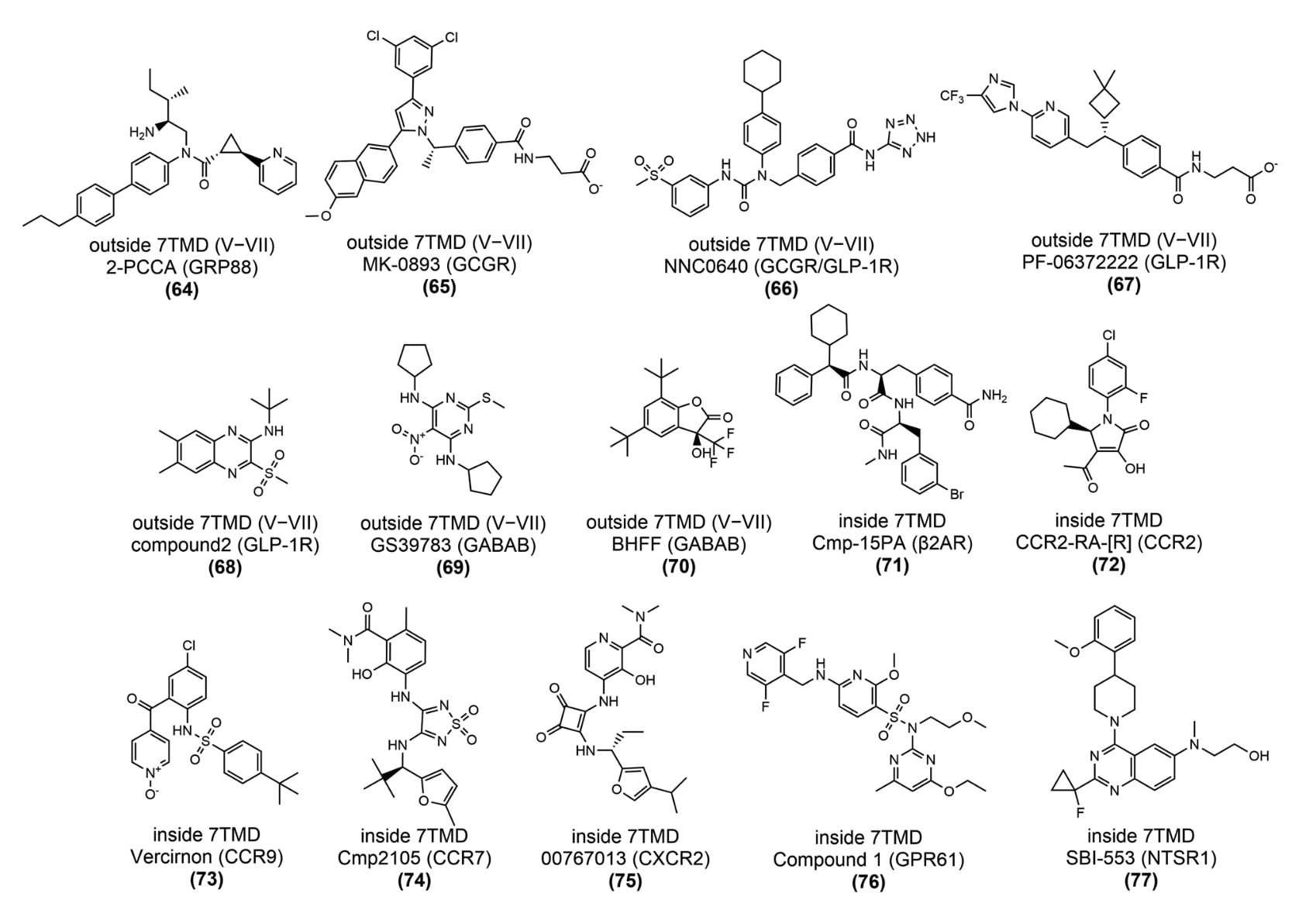
图32|展示了靶向GPCR胞内表面的小分子合成变构配体的二维化学结构。
- 7TMD内部:β2AR–Cmp-15PA、CCR2–CCR2-RA-[R]、CCR9–vercirnon、CCR7–Cmp2105、CXCR2–00767013、GPR61–Compound 1与NTSR1–SBI-553结构
不同于正构配体,GPCR可利用7TMD内部的胞内表面直接介导G蛋白或arrestin的下游信号转导。已解析结构显示,这里同样可作为变构调节剂结合位点,且各受体中该位点的结构高度相似,由TM I、TM II、TM VI与TM VII胞质端围成一个空腔。结合于此的变构拮抗剂可通过独特的双重机制抑制GPCR信号:一方面,它们通过占据G蛋白或arrestin的结合位点进行竞争;另一方面,这些分子还能与TM VII中保守NPxxY基序的Tyr7.53形成平行或T形π–π相互作用,充当“分子胶水”,将螺旋束胞质端锁定在一起,阻断活化相关的构象变化。因此,该药物化的变构口袋为开发GPCR小分子拮抗剂提供了新的可能。
趋化因子受体与配体在炎症反应中介导细胞趋化迁移。CCR9由CCL25激活,是炎症性肠病的重要靶点,因为其调控白细胞向肠道归巢。Vercirnon作为CCR9的变构拮抗剂,曾用于克罗恩病的临床开发,并进入Ⅲ期临床,但因需极高剂量才能有效阻断受体而表现出有限疗效。这可能源于分子进入细胞质后不一定能准确抵达G蛋白结合区域,这是此类调节剂共有的潜在挑战,目前仅vercirnon进入临床阶段。
晶体结构显示,vercirnon的结合位点位于7TM螺旋束胞内侧核心区域。其磺酰基与Glu3228.48、Arg3238.49与Phe3248.50的主链氨基形成三齿氢键,吡啶–N–氧化基与Thr81ICL1侧链形成氢键,而其酮基则与Thr2566.37侧链形成氢键。疏水相互作用方面,其叔丁基苯基深埋于由Val691.53、Val721.56、Tyr731.57、Leu872.42、Tyr3177.53与Phe3248.50组成的疏水口袋中;氯苯基则位于由Leu872.43、Ile1403.46、Val2596.40与Tyr3177.53形成的狭窄缝隙中。此外,吡啶–N–氧化结构被Thr81ICL1、Thr832.39、Asp842.40、Arg1443.50与Arg3238.49构成的极性腔体所包围。
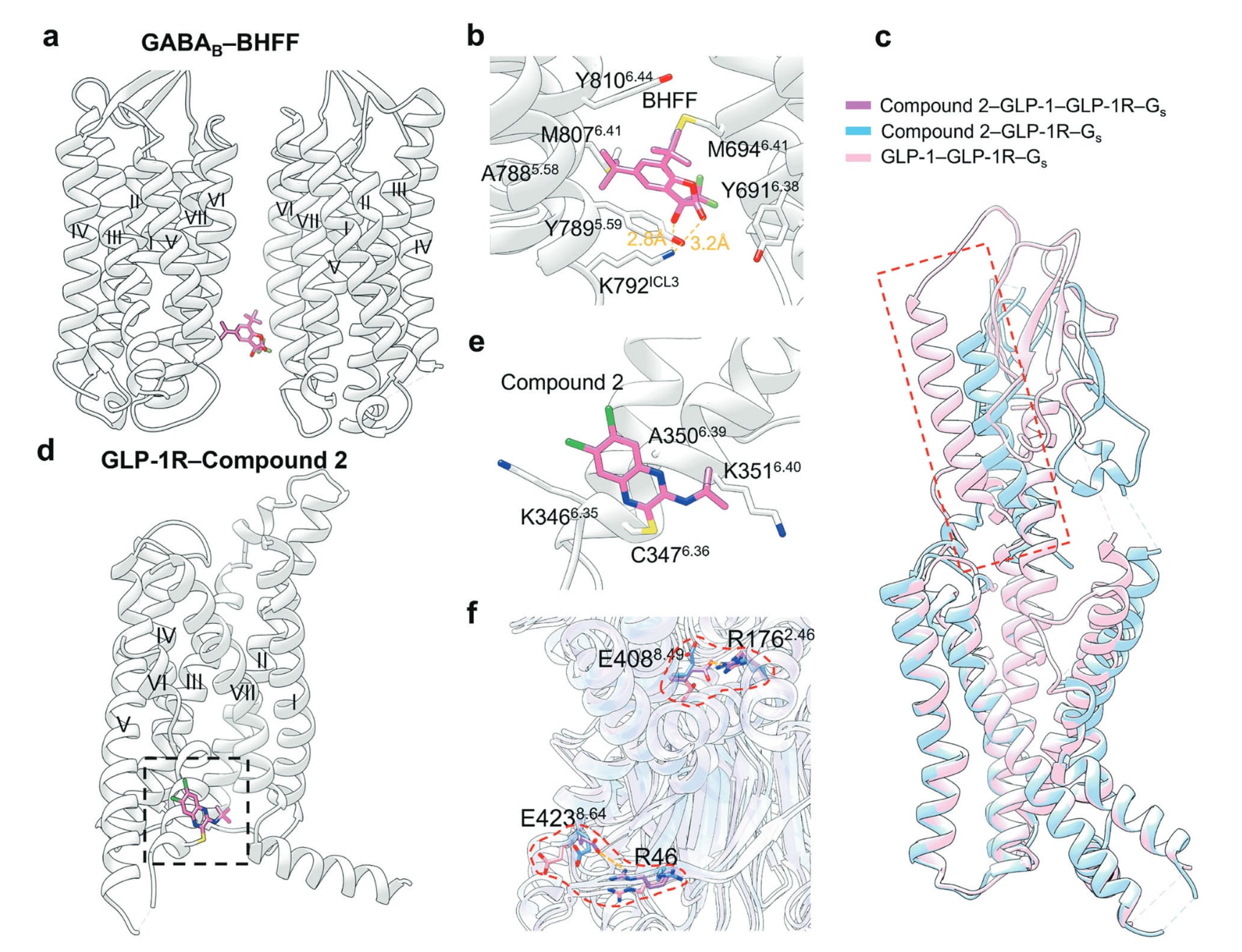
图33 a 展示了PAM BHFF与GABAB受体结合的示意图(PDB: 7C7Q)。b 描述了BHFF与GABAB受体的详细结合模式,其中氢键以橙色虚线表示。c 叠合展示了compound 2–GLP-1R–Gs结构(蓝色,PDB: 7DUR)与GLP-1–GLP-1R–Gs结构(粉色,PDB: 6×18)中N端α螺旋的构象差异。d 展示了ago-PAM compound 2与GLP-1R结合的示意图(PDB: 7EVM)。e 描述了GLP-1R结合compound 2的详细模式,包括疏水作用、共价键与极性相互作用。f 叠合呈现compound 2–GLP-1R–Gs(蓝色,PDB: 7DUR)、GLP-1–GLP-1R–Gs(粉色,PDB: 6×18)以及compound 2–GLP-1–GLP-1R–Gs(紫色,PDB: 7DUQ)结构中被重点标出的残基差异。
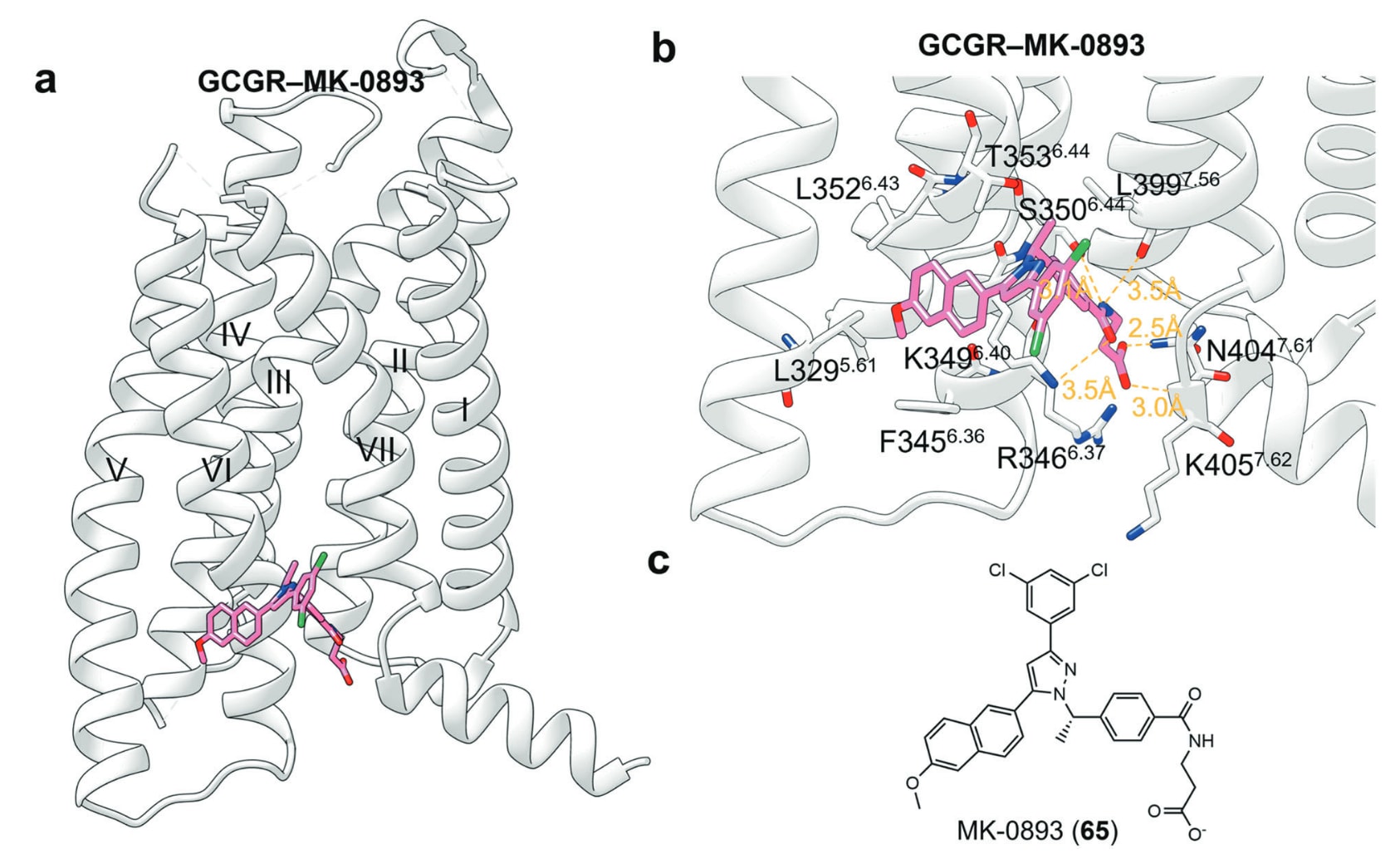
图34 a 展示了变构拮抗剂MK-0893与GCGR结合的示意图(PDB: 5EE7)。b 描述了MK-0893与GCGR的详细结合模式,氢键以橙色虚线标示。c 展示了小分子变构配体MK-0893的二维化学结构,用于辅助理解其构成与结合特征。
6 结论与展望
过去数十年中,基于晶体学、生物化学与计算手段的深入研究为GPCR结构调控带来了前所未有的原子级解析与结构理解。迄今已有388种正构调节剂被报道,并解析了717个与正构配体结合的GPCR复合结构。此外,还解析了53种不同的小分子变构调节剂及其九类变构结合位点。该研究首先回顾了GPCR的结构进展、信号机制与功能多样性,以勾勒该领域的整体格局与最新发展。通过上述结构学进展,正构与变构调节剂在其复合结构、信号作用机制以及药物研发中的启示方面得以分别讨论,强调了深入理解GPCR结构与机制对于开发有效治疗手段的重要性。
由于正构调节剂数量庞大且共享与内源配体竞争的功能机制,该研究特别选取并解析了近五年内具有代表性的正构药物案例,包括μ-OR–oliceridine复合物(以G蛋白偏向性信号见长)、S1PR–siponimod复合物(亚型选择性与“toggle switch”激活机制突出)、OX2R–lemborexant复合物(动力学与构象动态特征突出)、5-HT1F–lasmiditan复合物(以亚型选择性显著)以及GnRH1–elagolix复合物(基于受体非典型结构的独特信号传递方式)。结构分析揭示,正构调节剂通过与跨膜螺旋束内关键极性残基作用稳定在正构口袋中,其选择性往往由家族亚型之间非保守的关键残基决定。此外,“toggle switch”、PIF、DRY与NPxxY等基序是将胞外刺激传递至胞内的关键机械开关,而TM3与TM5/6之间特定极性作用亦可能强化TM5/6的位移,这是受体活化或去活化的核心标志。
鉴于变构调节剂的结合模式与作用机制往往高度独特,该研究特别强调了对变构调节剂的系统描述。结构分析显示,7TMD胞外前庭是最常见的变构结合位点,而诱导契合的构象适配与电荷匹配是变构配体进入口袋的关键。变构配体的结合会重塑受体的自由能景观,进而稳定不同的主导构象。基于此,该研究提出了全新的变构调节剂分类方式:第一类变构效应源于直接调控正构配体或胞内转导蛋白的结合,例如占据正构口袋上方并与正构配体或邻近残基直接作用以改变结合/解离,或直接占据G蛋白或arrestin的结合位点从而阻断其结合。第二类变构调节剂则通过间接改变受体活化路径、进而调控受体复合物与胞内转导蛋白的作用,它们往往以空间楔子的模式稳定或破坏特定相互作用,用以限制或促进受体构象重排。

图35 a 展示了变构拮抗剂vercirnon与CCR9结合的示意图(PDB: 5LWE)。b 描述了vercirnon与CCR9的详细结合模式,其中氢键以橙色虚线标示。c 展示了小分子变构配体vercirnon的二维化学结构,以便更清晰理解其化学骨架与结合特征。
除了前文总结的共性之外,从药物研发的角度进一步理解正构与变构调节剂的识别基础与精细机制,对于拓展药物设计边界具有重要意义。结构生物学与化学生物学的深入研究表明,理解GPCR的机械开关级联反应可帮助洞察精细调控策略,并使人类有能力控制GPCR的不同下游功能。基于此,经过突变改造的GPCR甚至可作为生物传感器以触发特定胞内信号。在此背景下,该研究提出五个对药物化学具有启发性的思路:
第一,正构小分子仍然是GPCR治疗的主体,因此高亲和力、高效能与高选择性的正构配体设计依旧是核心课题。对于提升亲和力与效能,可采用“anchor–driver”策略:其中“anchor”部分通过稳定结合主要残基提供亲和力,并在受体构象变化中保持相对不变;“driver”则设计成能与“toggle switch”等关键机械元件作用,从而触发受体活化或去活化。“anchor”提供基底,使“driver”能够通过“推”或“拉”驱动受体群体构象分布发生偏移,提高效能。这一“机制驱动的药物设计”理念拓展了传统“结构驱动药物设计”的边界。对于提升选择性,尽管正构口袋在亚家族内高度保守,但识别环区、跨膜束或关键残基间的微小差异并与这些热点区域特异作用,可显著提高选择性并减少副作用。
第二,寻找合适的变构口袋并进行变构药物设计仍是长期且充满挑战的研究方向。随着晶体学提供高可信度的初始结构,结合MD模拟与增强采样,可捕获受体中可能出现的隐匿变构位点。在识别这些口袋后,可运用三维分子生成方法基于拓扑表面与几何特征设计变构配体。尽管变构口袋保守性低,难以形成统一的骨架规律,但总体来讲疏水性仍是驱动结合的关键,而在7TMD外侧结合时往往还需极性基团帮助固定配体。此外,基于该研究总结的“类变构规则”(例如分子量≤600、可旋转键介于2–6、环数量≤5、最大环系由1或2个环组成、3≤SlogP≤7)可作为早期筛选标准。
第三,对于GPCR–配体复合结构已明确的体系而言,是否能有意识地调控配体的动力学与构象动力学性质值得重点考虑。以lemborexant优异的kon与koff参数为例,可推断:预先设计可在溶液中形成近似受体结合构象的配体有助于获得更高的kon,这可通过简单MD模拟筛选;而估算配体的结合自由能可用于预测并排序其koff,为动力学层面的优化提供指引。
第四,无论正构或变构调节剂,实现偏向性信号对于任何GPCR靶标都具有重要意义。以μ-OR的G蛋白偏向分子为典范,可通过比较偏向与非偏向配体的结合模式来识别导致偏向性的关键残基或区域,并以“机制驱动设计”为指导,通过构建或避免与这些热点的接触来实现信号的定向调控。
第五,在PAM与NAM调控机制中,正构与变构位点之间相互耦联。因此,将正构与变构药效团通过连接臂组合成的bitopic配体,有望深入探索位点耦联机制并获得高选择性与偏向性更强的分子。对于当前仍较为空白的GPCR bitopic领域,策略包括:在固定两端片段的前提下调控连接子;或在固定正构片段的基础上赋予变构片段“战斗部位”(warhead),使其锚定于含亲核残基的变构口袋。
尽管GPCR结构与药物研发已取得显著进展,但仍存在挑战。GPCR晶体学仍然耗时且繁琐,因此依赖结构的研究策略在一定程度上限制机理探索与药物发现,即便借助AlphaFold或RoseTTAFold仍存在残基取向不准确等问题,可能误导信号机制的解析。此外,MD模拟也无法保证识别所有潜在的隐匿变构位点。因此,序列、粗粒化拓扑与进化等新视角亟需引入,以减少对结构的依赖。其中一种可能性是利用庞大的序列数据与深度学习的端到端理念发展“sequence-to-mechanism”或“sequence-to-drug discovery”的策略,以避免在缺乏结构信息时模型累积误差。
综上,GPCR的正构与变构调控研究正处于深入发展阶段。虽然对小分子–受体之间作用、结合热点及调控机制的理解日益清晰,但仍有许多问题有待探索。该研究通过系统总结配体识别与调控机制,并提出新的变构调节剂分类与“机制驱动药物发现”理念,希望为该领域勾勒最新格局并激励进一步研究。未来,应致力于开发更高效、更具选择性且更安全的GPCR小分子治疗手段。