Annu. Rev. Pharmacol. Toxicol. 2014 | 受体别构位点的药物

Wenthur, C. J.; Gentry, P. R.; Mathews, T. P.; Lindsley, C. W. Drugs for Allosteric Sites on Receptors. Annual Review of Pharmacology and Toxicology, 2014, 54, 165–184. https://doi.org/10.1146/annurev-pharmtox-010611-134525.
0 摘要
在多种受体家族中发现具有可成药性且在拓扑结构上彼此不同的别构位点,为小分子调控受体功能提供了新的研究范式。与作用于正构位点的配体相比,靶向别构位点的配体在选择性方面具有显著优势,例如能够在同一受体家族内部实现亚型选择性,同时还可能赋予分子更优的理化性质。然而,别构配体并非解决所有问题的万能策略。随着研究的深入,一系列以别构调控为中心的化学与药理学问题逐渐显现,例如较为平坦的构效关系以及配体偏向性信号传导等。此外,由于别构位点在进化过程中保守性较低,这一特点虽然有助于提高选择性,但也可能带来物种之间的差异,从而在安全性评估中产生挑战。许多别构配体还表现出“分子开关”特征,即分子结构的细微变化(无论是化学修饰还是代谢变化)都可能改变其药理作用模式或受体亚型选择性。随着该领域不断发展,一系列关键原则与策略逐渐形成,用于指导针对别构位点的配体与药物设计。
1 引言
尽管小分子对蛋白质产生别构调控的概念早在20世纪中期就已被提出,但这一思想经过数十年的发展才逐渐受到广泛关注。许多研究将1965年视为别构理论正式确立的关键节点,因为Monod-Wyman-Changeux模型提出了“构象选择”机制,用以解释配体在细菌调控酶中的作用方式。然而,别构药物发现真正作为一种可行的治疗策略受到重视,则是在苯二氮卓类药物取得临床成功之后。该类药物属于别构配体,通过增强神经递质γ-氨基丁酸(GABA)在离子型GABAA受体上的作用而发挥药效,并避免了直接作用型GABAA受体激动剂可能带来的致命副作用。自这一发现以来,开发别构配体作为药物的研究兴趣持续增长。事实上,自1985年至今,关于别构受体调节剂的研究论文数量几乎呈指数式增长,这一趋势在专利文献中也同样明显。文献与专利活动的显著增加反映出别构配体研究已扩展至广泛的靶标类别,包括离子通道、激酶、半胱天冬酶、G蛋白偶联受体(GPCR)以及磷脂酶等。这些类别中包含大量具有治疗潜力的靶标,许多学术机构和商业团队均开展了相关研究。该领域的发展表明,要将别构配体成功开发为安全有效的治疗药物,需要新的小分子设计策略,并对药理学特性以及体内分布和代谢进行更加深入的评估。
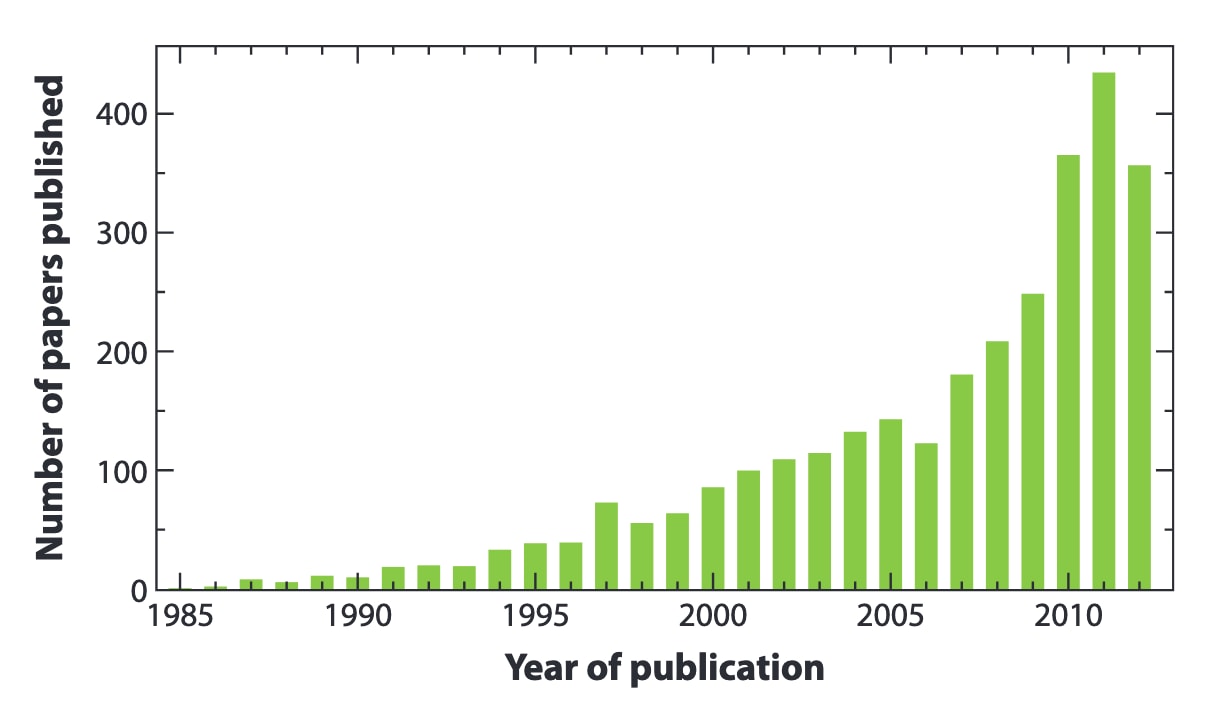
图1|根据Web of Knowledge数据库(http://www.webofknowledge.com/)统计,1985年至2012年间每年提及“别构受体调节剂”概念的文献数量。 美国专利商标局数据库(http://patft.uspto.gov/)中的相关趋势与之类似。
2 正构调控与别构调控
每一种受体或蛋白质通常都具有一个特定的内源性配体结合位点,该位点被称为正构结合位点(orthosteric binding site),而能够与该位点结合的天然或合成配体被称为正构配体。对于激酶而言,其正构配体为三磷酸腺苷(ATP);对于GPCR和离子通道而言,正构配体可能是小分子神经递质,例如谷氨酸或GABA,也可能是较大的多肽,如orexin。经典的合成正构配体通常通过放射性配体结合实验被发现,它们通过与内源性配体竞争结合靶点来发挥作用,并表现出多种药理效应,例如抑制剂、激动剂、拮抗剂或反向激动剂。 历史上,绝大多数获得美国食品药品监督管理局(FDA)批准的药物都作用于受体或蛋白质的正构位点。然而,这类配体往往存在疗效不足、长期用药后疗效下降、选择性有限或耐药性等问题。
在GPCR、离子通道、半胱天冬酶、激酶和磷脂酶等多类蛋白中,除了正构结合位点之外,还存在拓扑结构和功能上均与之不同的别构结合位点。别构位点的存在使得配体与受体之间能够形成更多样的相互作用方式,并产生不同于正构位点调控的信号调节现象。高通量筛选(HTS)技术和功能性实验方法的发展进一步推动了这一研究方向,使得能够识别出影响靶标功能但不必局限于正构位点结合的小分子。尽管不同靶标的药理特征各有差异,但对于具有构象动态变化特性的受体或蛋白质,小分子可以通过结合别构位点来稳定其特定构象,无论是在内源性正构配体存在或不存在的情况下均可发挥作用。当别构配体稳定蛋白质的活性构象时,会引发受体激活与信号传导;若稳定其非活性构象,则会阻断信号。由于别构配体能够稳定蛋白质的一种独特构象,蛋白质与配体形成的复合物实际上可以被视为一种新的受体状态,从而表现出独特的药理特性,例如配体偏向性信号传导。
在GPCR、激酶和离子通道等靶标中,由于正构位点或ATP结合位点高度保守,同时许多合成正构配体在理化性质以及药物代谢和药代动力学方面存在不利特征,因此开发具有高度选择性的正构化合物往往困难重重。 在许多情况下,直接作用型激动剂还可能产生毒性,或者由于长期持续激活受体而导致受体脱敏、内吞或下调。相比之下,别构配体通常结合于在进化上保守性较低的位点,因此往往能够实现更高水平的选择性。此外,别构配体具有“天花板效应”,即当别构位点被完全占据后,药效不会继续增加。同时,如果某种别构调节剂本身不具有激动剂活性,它仅在内源性激动剂存在时才发挥作用,从而能够保持内源性配体在时间和空间上的生理调控特征。多项研究还表明,与正构配体相比,别构配体在化学可优化性以及理化性质方面往往更具优势。
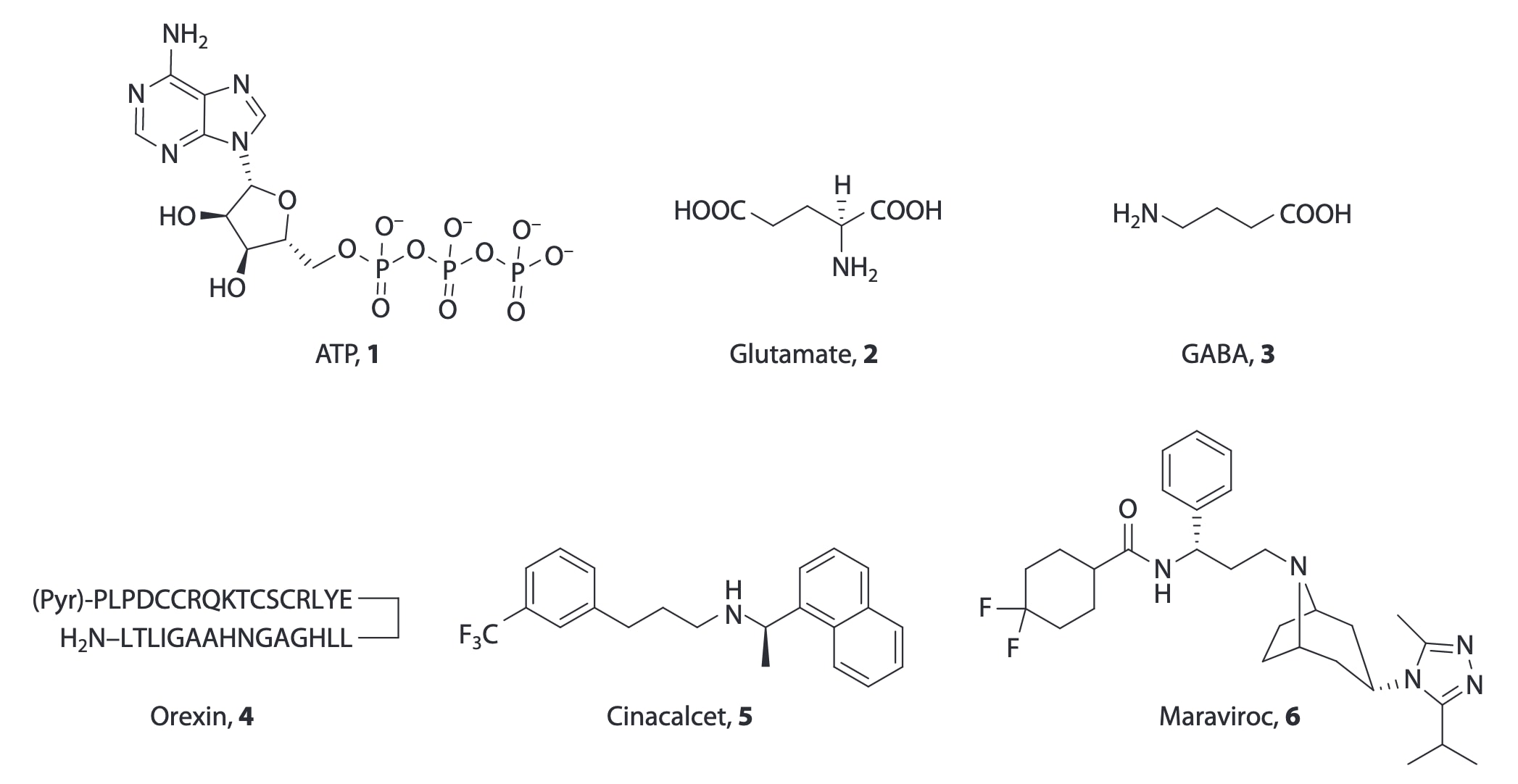
图2|内源性正构配体1–4以及已上市别构配体5和6的结构。 缩写:ATP,三磷酸腺苷;GABA,γ-氨基丁酸。
在GPCR和离子通道体系中,别构配体可以表现出多种不同的药理模式。其中包括正向别构调节剂(PAM),其能够增强激动剂诱导的受体反应;负向别构调节剂(NAM),其通过非竞争方式降低受体活性。此外,还存在沉默型别构调节剂(SAM,也称中性别构配体),这类配体同样结合别构位点并可阻断PAM或NAM的作用,但本身不会改变正构配体引发的受体反应。研究还发现更加复杂的别构调控模式。例如,部分研究致力于开发别构激动剂,即在没有正构配体存在时即可激活受体的别构化合物。然而,许多所谓的别构激动剂实际上可能是双位点配体(bitopic ligand),即同时结合正构位点与别构位点的杂合配体。这类配体往往表现出依赖受体表达水平的药理行为、配体偏向性信号传导以及复杂的构效关系。此外,一些别构配体表现为部分拮抗剂,即完全占据NAM位点但只能部分阻断受体信号;还有一些被称为ago-PAMs,即既具有PAM作用又具有一定别构激动活性的分子。然而,在同一化学系列中,这些药理模式往往变化较大,使得构效关系分析变得更加困难。
对于激酶和磷脂酶而言,别构配体通常结合于远离正构(催化)位点的远端区域,并稳定蛋白质的特定非活性构象。靶向这些位点在亚型选择性、激酶组选择性以及安全性方面均展现出显著优势。尽管别构调控相较于正构调控具有诸多优势,但其并非药物发现的万能策略。在开发别构配体时仍需面对多方面的药理学与化学挑战。从药理学角度看,主要问题包括:别构位点较低的进化保守性可能导致显著的物种差异;别构调节剂的状态依赖性在某些伴随内源性正构信号逐渐减弱的退行性疾病中可能成为不利因素;别构调节剂引入的信号偏向性可能产生不期望或难以预测的生理效应;此外,别构调节剂还可能同时作用于受体的同源二聚体和异源二聚体,从而使生理反应更加复杂。从化学角度看,主要困难包括别构配体往往具有较为平缓的构效关系、难以引入极性或增溶基团(从而降低logP值),以及分子开关现象的存在。这种分子开关既可能由细微的化学结构变化引发,也可能由体内氧化代谢产生,因此需要更加深入的分子药理学研究。
尽管存在这些挑战,已有两种GPCR别构调节剂成功上市,分别是钙敏感受体的正向别构调节剂cinacalcet以及CCR5受体的负向别构调节剂maraviroc。 此外,许多别构激酶抑制剂正处于不同阶段的人体临床试验中,而苯二氮卓类药物作为调控离子通道的别构调节剂,也已成为非常成功的治疗药物。
3 别构调节剂的治疗靶点
作为临床前研究中的工具化合物和探针,别构配体已经针对多种治疗相关靶标被开发,包括离子通道、半胱天冬酶、激酶、磷脂酶以及G蛋白偶联受体(GPCR)。针对任何一个靶标类别的深入讨论都足以占据整篇文章的篇幅,并且已有大量综述对这些靶标及其别构配体进行了详细总结。因此,该节主要讨论激酶和磷脂酶,而下一节将重点介绍GPCR别构配体设计所呈现出的复杂性。
3.1 利用别构调节剂靶向激酶
蛋白质磷酸化是细胞信号传导中的一种关键调控方式,几乎存在于所有已知的细胞信号通路中。激酶活性的异常与多种人类疾病密切相关,包括糖尿病、癌症以及多种神经系统疾病,因此激酶被认为是极具吸引力的治疗靶标。近年来美国食品药品监督管理局(FDA)批准的激酶抑制剂数量持续增加,也反映出制药企业在该领域的高度关注。然而,尽管已经取得了一些进展,激酶仍然是较难开发的靶标,这主要源于人类激酶组之间高度保守的结构特征。这种结构保守性来自激酶共同的催化功能,即将捕获的ATP中的γ-磷酸基转移到蛋白质底物上。 ATP结合口袋在结构上高度保守,而许多早期开发的激酶抑制剂正是作用于该位点,因此在500多种已知激酶之间往往缺乏选择性。为了获得更具选择性的化合物,研究者开始寻找位于ATP结合口袋之外、可被小分子靶向的结合位点。
与ATP竞争结合的激酶抑制剂通常被称为I型抑制剂。 进一步研究发现,还存在三类额外的别构抑制剂,其区别主要在于作用位点的不同。 II型抑制剂结合于ATP位点并延伸至邻近的别构口袋;III型抑制剂则仅结合于靠近ATP位点的别构口袋。 这两类抑制剂通过将激酶锁定在非活性构象来实现抑制作用,使高度保守的激活环被迫离开ATP位点附近的疏水口袋。 相比之下,IV型抑制剂则结合于距离ATP位点更远的别构区域。
目前已报道的别构激酶抑制剂表现出多种作用机制,例如诱导蛋白结构重排、阻止活性复合物形成、阻断底物识别、稳定非活性复合物、诱导蛋白降解或占据自抑制位点等。然而,有证据表明许多别构抑制剂实际上倾向于稳定少数几种在激酶中反复出现的非活性构象。特别是作用于若干已知别构口袋的配体,例如MEK口袋、Akt1口袋、PIF口袋以及ANS口袋,被认为通过破坏与附近高度保守的αC螺旋之间的相互作用而实现抑制作用。此外,MEK口袋和PIF口袋在多个激酶家族中均被发现,并在同一激酶家族成员之间产生类似的调控效应。
由于激酶在治疗中的重要性,靶向这些保守性较低且能够稳定非活性构象的别构位点已成为激酶药物开发的重要策略。针对Akt、Abl、PDK1、JNK1、CHK1、IGF-1R、CDK2以及mTOR等激酶的别构抑制剂已经被开发出来,而且它们在结构上通常与ATP竞争性抑制剂几乎没有相似性。其中,针对Akt激酶的别构抑制剂代表了一种新的激酶抑制机制,并且已进入II期临床试验阶段。Akt包含三种人类同工酶Akt1、Akt2和Akt3,在细胞质中通常以PH-in闭合构象存在,此时pleckstrin homology(PH)结构域阻挡ATP结合口袋以及T308/S473位点。当Akt被招募到细胞膜后,激酶转变为PH-out构象,从而暴露ATP结合口袋并允许其被磷酸化。
Merck研究团队开发了一类依赖PH结构域的小分子Akt抑制剂,这些化合物对同工酶具有选择性,并且不与ATP竞争,也不会抑制激酶组中的其他成员。有趣的是,别构抑制剂7不仅能够抑制Akt的激酶活性,还能够抑制Akt自身的磷酸化。大量的生化实验、突变研究以及构效关系分析表明,别构抑制剂7以及随后开发的临床候选药物MK-2206能够稳定Akt的PH-in闭合构象。随后X射线晶体结构研究证实了这一机制:别构抑制剂7结合于由PH结构域与激酶结构域界面残基形成的疏水口袋中,并与Trp80形成π-π堆积作用,同时在蛋白两个结构域中形成氢键。进一步的体内荧光共振能量转移实验表明,该抑制剂能够将Akt锁定在PH-in构象,从而阻止T308和S473位点的磷酸化。
这一研究也进一步凸显了别构抑制剂的优势。例如,ATP竞争性Akt抑制剂(如化合物9和10)在结合ATP位点后会诱导Akt发生过度磷酸化,从而引发对靶标的调控性磷酸化。相比之下,别构Akt抑制剂(如7和8)不仅能够抑制Akt的生理性激活,还能够抑制药物诱导的Akt过度磷酸化,从而扩大治疗窗口,并可能提高MK-2206的临床疗效和耐受性。
该领域的发展在一定程度上受到高通量筛选技术不足的限制,因为识别能够稳定非活性构象的别构激酶抑制剂仍然较为困难。过去通常需要通过耗时且复杂的ATP竞争实验作为二级筛选手段,以识别非竞争性的别构配体。近年来,一代新的结合实验技术开始用于检测能够稳定激酶构象的配体。其中FLiK(kinase荧光标记技术)能够在不依赖激酶活性的情况下检测配体诱导的构象变化,例如在p38α和Abl激酶中。然而,即使有了这些技术进展,仍然需要更多新的筛选方法和实验体系,以更高效地在大规模化合物库中发现能够通过复杂别构机制调控激酶活性或磷酸化的小分子配体。
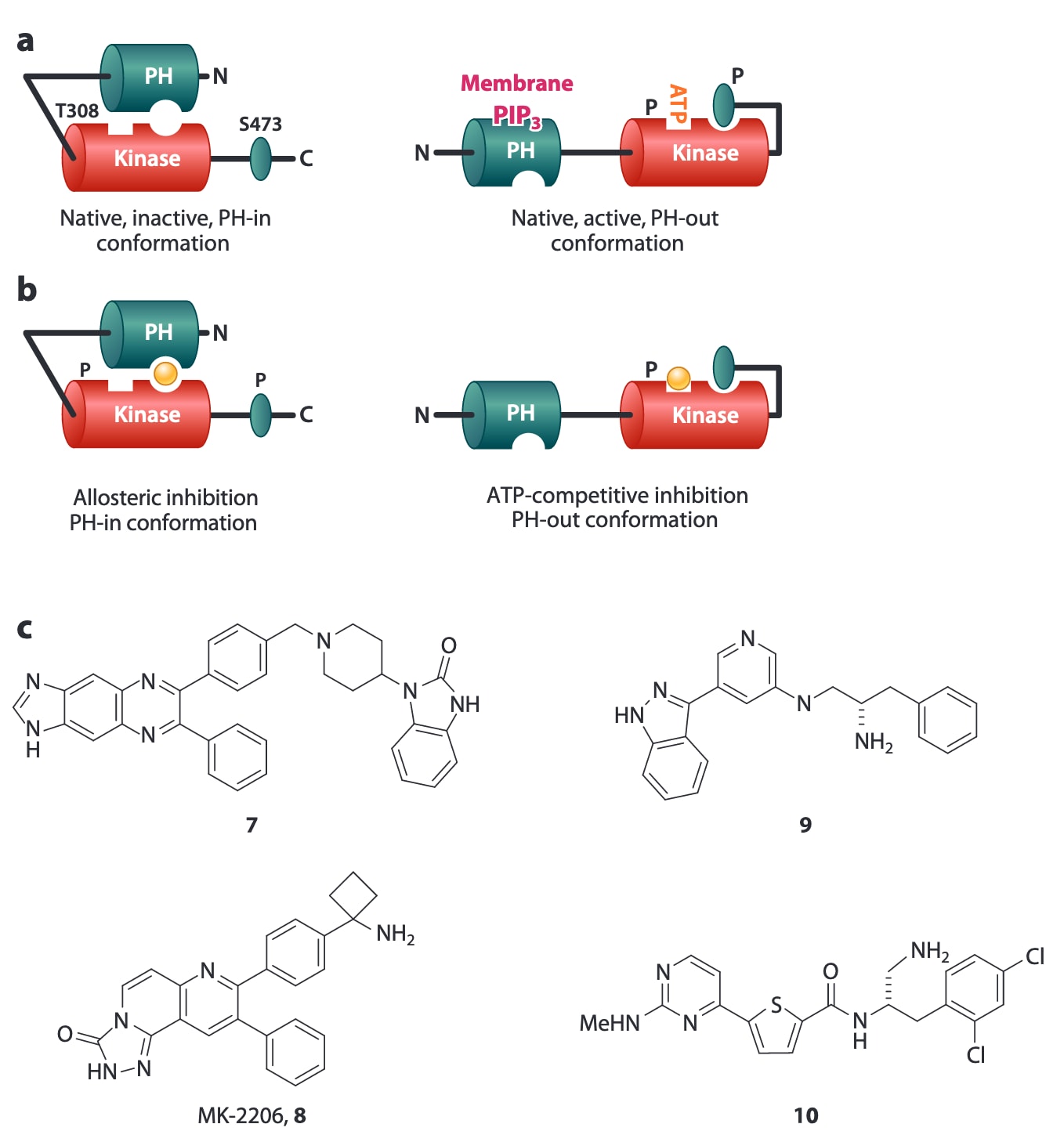
图3|构象具有柔性的Akt蛋白。 (a) 细胞质中处于非活性状态的Akt构象,也称为PH-in构象。当受到PIP3招募至细胞膜时,激酶转变为活性的PH-out构象,从而暴露T308和S473位点以进行磷酸化。(b) 别构抑制剂7和8对Akt的抑制作用会使其保持在PH-in构象,从而同时抑制激酶活性和磷酸化。ATP竞争性抑制剂结合于催化位点,使Akt保持PH-out构象。尽管激酶活性受到抑制,但由于这种构象状态,Akt仍可能发生过度磷酸化。(c) 别构Akt抑制剂7和8以及ATP竞争性Akt抑制剂9和10的结构。缩写:ATP,三磷酸腺苷;PH,pleckstrin同源结构域;PIP3,磷脂酰肌醇(3,4,5)-三磷酸。
3.2 利用别构调节剂靶向磷脂酶
调节酶活性的小分子化合物通常可以分为两大类:竞争性抑制剂和别构抑制剂。竞争性抑制剂通常被称为基于底物的抑制剂,其作用方式是靶向酶或受体中用于结合天然底物或配体的位点。这类抑制剂之所以被称为“竞争性”,是因为其结构往往模拟天然底物或配体,从而与其竞争结合。相比之下,别构抑制剂则结合于酶或受体表面上与底物结合域不同的位点。这种别构结合机制通常可分为两种形式:非竞争性抑制和反竞争性抑制。非竞争性抑制剂会在底物结合之前结合于酶的另一个位点,从而改变活性位点构象,使天然底物无法结合;而反竞争性抑制剂则与酶-底物复合物(ES复合物)结合,从而阻碍产物形成。竞争性抑制与别构抑制的作用机制可以通过酶抑制动力学特征加以区分。
基于底物的抑制剂通常模拟酶的底物,因此抑制剂
竞争性抑制和别构抑制在药物设计中各自具有不同的优势与局限。然而,在脂质代谢酶的小分子抑制剂设计中,别构结合机制具有明显优势。酶-底物复合物的形成由体系自由能变化决定,可表示为
其中
随着脂质信号网络在疾病中的重要性不断提高,亟需开发能够阐明特定酶同工型在信号脂质生成过程中作用的化学工具。近年来,磷脂酶逐渐成为备受关注的潜在药物靶标。磷脂酶是一类催化磷脂水解反应的酶,根据催化反应类型可分为四大类:磷脂酶A(包括A1和A2)、磷脂酶B、磷脂酶C以及磷脂酶D(PLD)。PLD是一种脂质信号酶,能够催化磷脂酰胆碱水解生成磷脂酸这一重要的脂质第二信使以及胆碱。研究已经鉴定出两种哺乳动物PLD同工酶,即PLD1和PLD2,两者序列同源性约为53%,但在功能上存在差异。两种同工酶都具有保守的组氨酸-赖氨酸-天冬氨酸催化结构域,同时在N端具有保守的PX结构域和PH调控结构域。
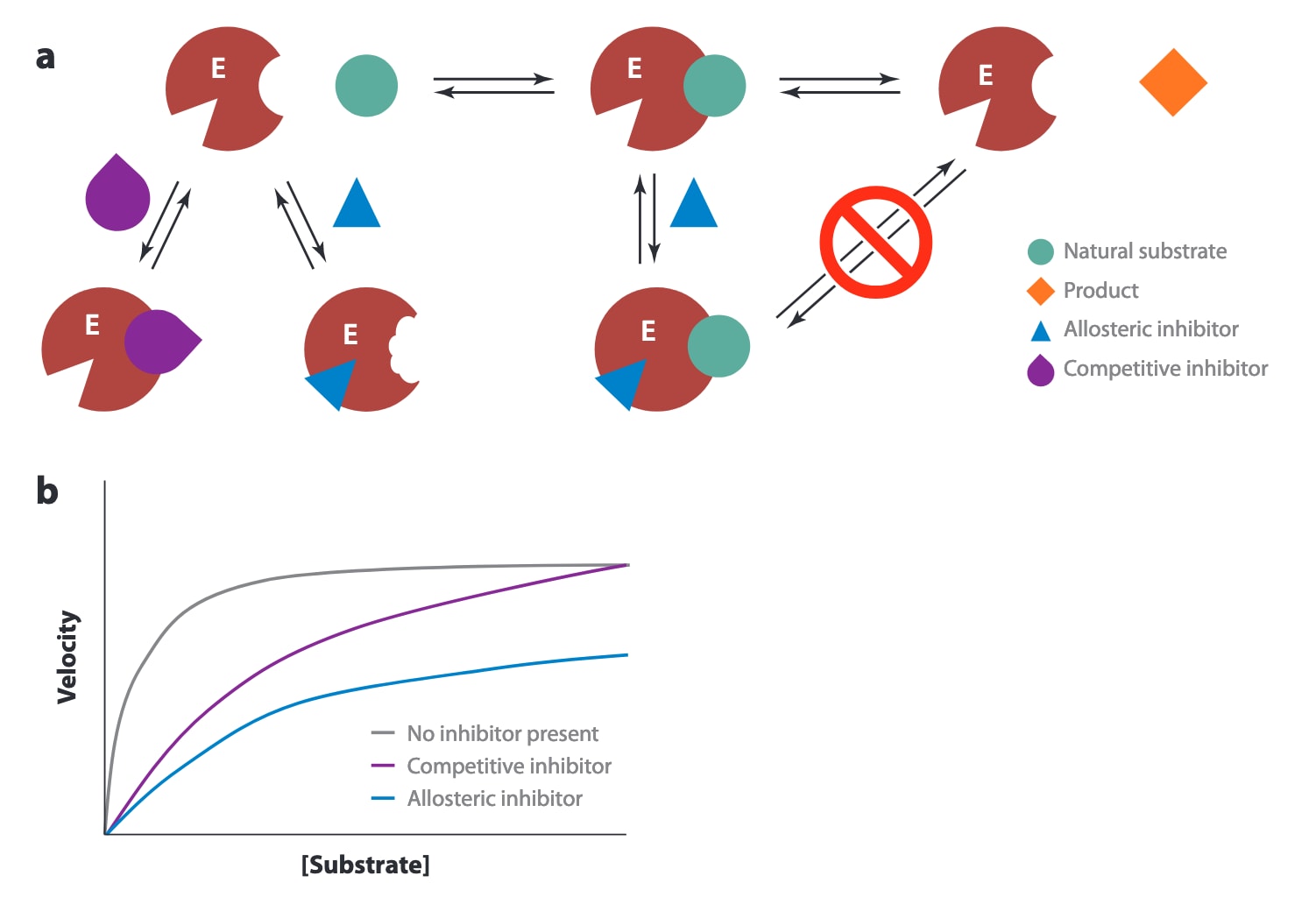
图4|别构抑制剂与基于底物的抑制剂。 (a) 调节酶活性的小分子化合物通常可分为两大类:竞争性抑制剂(也称为基于底物的抑制剂)和别构抑制剂。基于底物的抑制剂(紫色)结合于酶的活性位点,从而阻止天然底物被催化消耗。别构抑制剂(蓝色)则结合于酶表面不同的位点,以阻止底物结合(该过程称为非竞争性抑制),或阻止在酶-底物-抑制剂(ESI)复合物中产物的形成(该过程称为反竞争性抑制)。(b) 这两种机制可以通过动力学分析加以区分。基于底物的抑制剂会提高酶对底物的表观
PLD功能失调与多种疾病相关,包括癌症、中枢神经系统疾病以及病毒感染过程中的关键阶段。然而,长期以来用于抑制PLD活性的工具主要局限于遗传学和生化方法,例如使用n-丁醇这一在转磷脂酰化反应中与水竞争的配体。2007年发现20世纪80年代开发的抗精神病药物halopemide能够抑制PLD活性,是该领域的重要突破。Halopemide是一种多巴胺受体拮抗剂(D2
基于PLD酶的构象柔性、PH结构域的存在以及halopemide所含的哌啶苯并咪唑酮结构,该化合物在结构上与能够产生别构Akt激酶抑制作用的化学类型相似。因此,halopemide成为研究PLD抑制模式并开发同工酶选择性抑制剂的重要先导化合物。通过多样性导向合成策略构建了约260个类似物,其中VU0359595被发现是一种对PLD1具有1700倍选择性的抑制剂。研究表明,苯并咪唑酮核心结构有利于PLD1抑制,而乙二胺连接臂上的手性(S)-甲基基团进一步增强了PLD1抑制活性。另一方面,通过采用生物等排体三氮杂螺酮骨架,可以获得优先抑制PLD2的化合物VU0364739,其对PLD2的抑制选择性比PLD1高约75倍。VU0359595和VU0364739以及该系列更为优化的PLD抑制剂在药物代谢和药代动力学性质方面均显著改善,同时消除了生物胺相关活性。
有趣的是,当将手性(S)-甲基基团引入PLD2选择性的三氮杂螺酮骨架后,PLD1活性提高了150倍以上,从而得到能够同时强效抑制PLD1和PLD2的化合物。这一现象构成了磷脂酶配体中首个“分子开关”实例。针对缺失N端结构域(靠近PX和PH结构域)的PLD突变体研究表明,其活性明显降低。结合其他生化实验数据,可以确认VU0359595和VU0364739以高亲和力结合于PH结构域中的某个位点,并诱导蛋白构象变化,从而在酶的其他区域形成第二个结合位点。这种结合特征与Akt别构抑制机制类似,并能够实现前所未有的PLD同工酶选择性。
像VU0359595和VU0364739这样的化学工具在解析不同PLD同工酶在多种生物体系中的作用方面发挥了重要作用。与激酶别构调节剂类似,磷脂酶的别构调节剂也难以通过现有筛选方法和技术被直接识别。
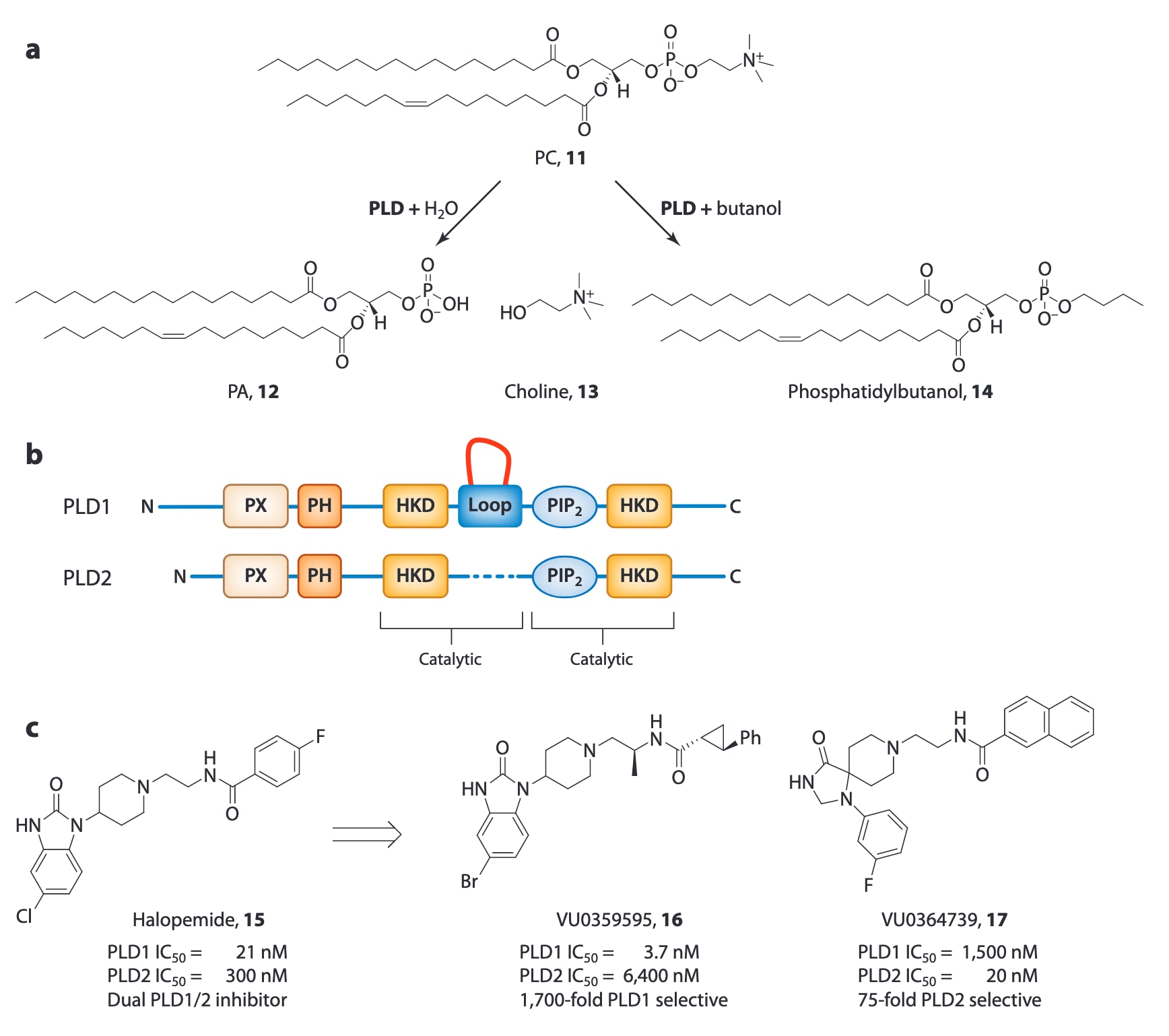
图5 (a) PLD的生物化学反应。PLD催化磷脂酰胆碱PC(11)水解生成磷脂酸PA(12)和胆碱(14)。在存在一级醇(如
4 GPCR别构配体设计日益显现的复杂性
近年来的多篇综述已经系统总结了GPCR别构配体在药理学和药物开发中的注意事项,并列举了大量已知的GPCR别构调节剂。在所有别构配体类型中,调控GPCR功能活性及其下游信号通路的配体研究最为成熟,相应的动力学与功能性检测方法(如钙离子或铊离子通量实验)也已较为完善,可用于高通量筛选以及常规筛选流程,例如三重加样(triple-add)实验体系。正如前文所述,GPCR别构调节剂通常表现出较为平缓的构效关系(SAR),即使是配体结构中细微的电子效应或空间效应变化,也可能导致活性完全丧失,从而显著增加化学优化以及药物代谢和药代动力学性质优化的难度。该节将讨论近年来关于分子开关、ago-PAMs、别构激动剂(更准确地说为双位点配体)以及配体偏向性信号传导的新认识与相关问题。
4.1 分子开关
在别构理论的发展过程中,人们逐渐认识到,在三元复合物模型中,别构化合物的活性主要由两个因素决定:配体的亲和力以及协同作用。随后提出的激动剂作用操作模型进一步指出,这两个因素可以使受体复合物表现出独特的效能。换言之,某些别构配体无论单独存在还是与其他配体共同存在,都可能对靶标具有较高的结合亲和力,但却不会对整体复合物的信号传导性质产生明显影响。当这一现象与蛋白质中微小构象变化即可显著影响信号功能这一事实结合时,就产生了一种可能性:一系列结构非常相似的化合物可能同样能够有效结合靶标,但各个成员却会表现出完全不同的药理模式。事实上,这种现象已经在多种化学系列以及多类蛋白靶标中被反复观察到。
在同一化学系列中,结构相近的化合物可能分别表现为激动剂、正向别构调节剂(PAM)、负向别构调节剂(NAM)或沉默型别构调节剂(SAM),这种现象被称为作用模式转换(mode switching),而导致这些差异的细微结构变化通常被称为分子开关。该现象在代谢型谷氨酸受体5(mGlu5)的研究中被频繁报道,不同研究团队在多个与2-methyl-6-(phenylethynyl)pyridine(MPEP)别构结合位点相关的化学骨架中均观察到了这种分子开关现象。这一现象并不限于mGlu5,在II类和III类代谢型谷氨酸受体中也有类似报道。事实上,作用模式转换似乎在多种别构靶标中普遍存在,包括毒蕈碱型受体、趋化因子受体以及电压门控钾离子通道等。
尽管相关研究积累了大量构效关系数据,但对于导致作用模式转换的配体-受体相互作用机制,目前仍缺乏统一的解释。近年来,对沉默型别构调节剂(SAM,也称为中性别构配体)的研究为理解别构结合位点提供了重要工具,也在一定程度上促进了对亲和力与协同作用分子决定因素的认识,尤其是在mGlu受体研究中。SAM通常在放射性配体结合实验中表现出较高的别构位点亲和力,但在功能实验中不表现出明显效能,因此可用于与已知PAM或NAM进行竞争分析。然而,一种在某种功能实验中表现为SAM的化合物,在另一种功能实验中可能表现出活性,这通常与配体偏向性信号传导有关。此外,由于结构相似的化合物常在不同受体亚型之间表现出不同的活化模式,一些具有亚型选择性的化合物在相关受体上可能表现为SAM。这些复杂情况表明,需要在多种功能实验体系以及相关受体的结合实验中系统评估别构调节剂的活性,才能获得对其药理作用更加准确的认识。
除了利用SAM进行研究外,定点突变实验也被证明是揭示受体关键相互作用位点的重要方法,可用于区分导致正向或负向协同作用的结构因素。例如,在mGlu5受体的研究中,通过突变关键氨基酸残基,可以使原本的PAM转变为NAM,反之亦然。类似的方法也被成功应用于促甲状腺激素受体研究,为激素受体别构配体的理性设计提供了可能的结构基础,也证明了在研究作用机制时引入突变实验的重要价值。
在讨论作用模式转换时,还需要考虑代谢过程对别构调节剂药理特性的影响。由于结构极其相似的化合物可能表现出完全不同的药理模式,因此某些别构配体的代谢产物也可能通过形成分子开关而产生与母体化合物相反的作用。近期报道了首个由细胞色素P450(CYP)介导的分子开关实例。在该研究中,一种mGlu5 PAM化合物VU0403602在体外实验中不具有激动剂活性,但在体内却诱发了典型的mGlu5激动剂样癫痫样活动。这种体内效应可以被MPEP阻断,表明其与mGlu5有关,同时也可以被泛CYP抑制剂ABT(1-氨基苯并三唑)抑制。进一步的代谢研究发现,VU0403602经氧化代谢产生的代谢物M1实际上是一种强效的mGlu5别构激动剂,并由其引发了体内观察到的癫痫样活动,而非母体化合物本身。在药物研发过程中,如果某一代谢产物具有与母体化合物完全不同甚至具有毒性的药理特性,将对临床开发带来重大风险,因此需要更加重视别构配体主要代谢物的药理学特征。
最后,分子开关不仅可以改变药理作用模式,还可能影响受体亚型选择性。在某些情况下,这种变化可能带来不利影响,而在其他情况下,则可能使研究者获得此前难以获得的亚型选择性配体。近期的一项研究利用这一特性成功开发出首批高度选择性的mGlu3 NAM。在该研究中,一种强效mGlu5 PAM化合物VU0092273在mGlu3上仅表现出较弱活性。随后通过并行迭代合成寻找能够消除mGlu5活性并增强mGlu3活性的分子开关。结果发现,在南侧苯环上引入
该研究还同时解决了平缓SAR带来的问题。mGlu3 NAM VU0469942在大鼠体内清除率较高,其主要代谢途径是CYP介导的
4.2 Ago-别构调节剂
Ago-别构调节剂是指一类能够通过别构位点结合的配体,这类配体在没有正构配体存在时即可诱导受体产生反应,同时在正构配体存在时又能够增强其效应。此类配体的发现与鉴定过程通常较为复杂,因此需要将放射性配体结合实验与多种功能实验结合使用。在许多研究中,一种别构配体在进一步研究以及筛选技术不断发展的背景下,往往经历从最初分类到重新分类,甚至多次重新分类的过程。
例如,最早报道的ago-别构调节剂之一是2-氨基噻吩类化合物PD81,723。最初的研究认为该化合物是腺苷A1受体的纯正向别构调节剂(PAM),因为它能够增强
尽管ago-别构调节剂的鉴定与表征过程较为复杂,但对这类化合物及其作用机制的研究兴趣仍在不断增加。Noetzel等人近期比较了mGlu5受体ago-PAM化合物VU0360172和VU0092273与纯mGlu5 PAM在中枢神经系统中的功能差异。在大鼠体内实验中,这两类化合物在逆转安非他命诱导的过度运动方面表现出相似的效果,而该效应通常被用作潜在抗精神病活性的预测指标。此外,研究还在表达mGlu5受体的不同细胞系中分析了VU0360172和VU0092273的体外作用。这些细胞系的受体表达水平不同,实验结果显示,在受体表达量较高的细胞系中,这些化合物表现为ago-PAM,而在受体表达量较低的细胞系中则表现为纯PAM。这些结果表明,在体外实验中评估ago-别构调节剂时需要充分考虑受体表达水平的影响,同时也提示在天然生理体系中,由于不同脑区受体储备水平可能存在差异,现有实验方法未必能够完全准确地区分纯PAM与ago-PAM在实际生理系统中的效应。
别构调节剂长期以来被认为具有开发为新型治疗药物的潜力,其优势包括较高的受体亚型选择性以及较低的受体脱敏或内吞倾向。然而,这类配体通常依赖于内源性配体的存在才能发挥作用。因此,ago-别构调节剂所具有的内在激动剂信号特性,在某些情况下可能成为一种有吸引力的治疗策略,例如在退行性疾病中,随着疾病进展,内源性配体水平逐渐降低,或者在某些突触中内源性信号本身较弱。然而,在其他情况下,例如mGlu5 PAM的开发中,研究通常更倾向于获得纯PAM,因为ago-PAM可能会诱发癫痫样活动。
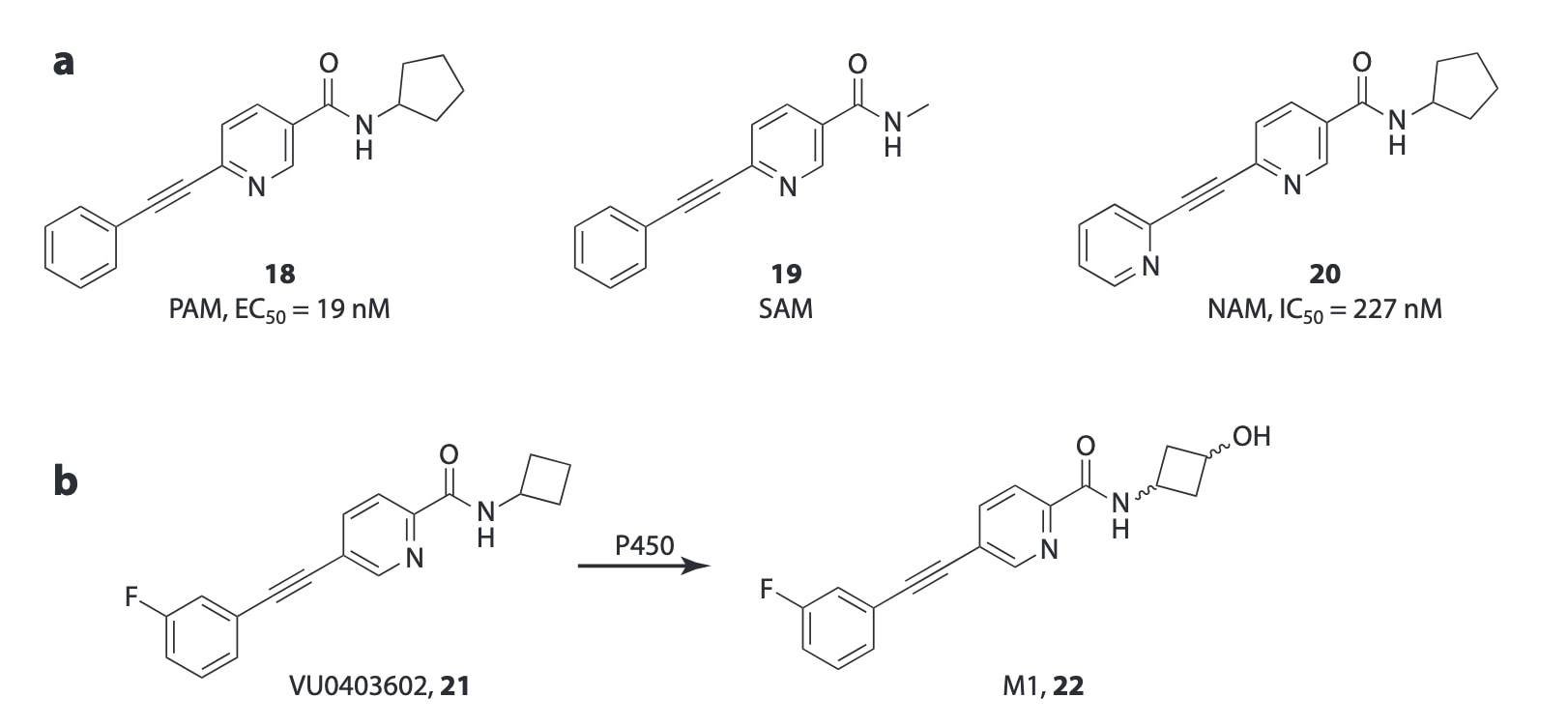
图6 (a) MPEP位点mGlu5别构配体系列中分子开关的经典实例。一个强效mGlu5 PAM(18)通过将环戊基酰胺替换为甲基酰胺转变为mGlu5 SAM(19)。相反,在PAM(18)的南侧苯环中引入一个氮原子后,得到吡啶衍生物(20),其表现为mGlu5 NAM。(b) mGlu5 PAM VU0403602(21)在体内代谢生成具有mGlu5别构激动剂活性的代谢物(22),从而导致毒性。缩写:mGlu5,代谢型谷氨酸受体5;MPEP,2-methyl-6-(phenylethynyl)pyridine;NAM,负向别构调节剂;PAM,正向别构调节剂;SAM,沉默型别构调节剂。
4.3 别构激动剂
在前文讨论PAM与ago-PAM时提到的受体储备依赖性药理学问题,同样适用于别构激动剂的研究。别构激动剂是指一类结合于不同于正构位点的别构位点、并在缺乏内源性配体时即可激活受体的配体。GPCR研究领域对这一策略的关注显著增加,部分源于Portoghese将此类配体应用于阿片受体二聚体研究,并证明这些配体相较于单价配体可能具有更高的结合亲和力以及更好的亚型选择性。近年来,双位点配体(bitopic ligands)的研究进一步利用了这些潜在优势,并结合GPCR丰富的别构药理学特性展开发展。
双位点配体在结构上类似于异源双价配体,它们通常由两个不同的药效团通过一段适当长度且具有一定柔性的连接臂共价连接而成。但与传统双价配体不同的是,双位点配体中的一个药效团作用于正构位点,而另一个作用于别构位点,因此能够同时靶向同一受体的正构与别构位点。由于具有这种独特的正构-别构组合特征,双位点配体往往表现出复杂的结合行为,其机制可能介于别构三元复合物模型与正构竞争结合模型之间。目前提出的结合模式主要包括三种情况:双位点配体同时占据正构位点与别构位点;双位点配体仅占据其中一个位点,即所谓的“翻转”(flip-flop)结合模式;或者两个配体以协同方式分别占据这两个位点。
将不同药效团组合以产生协同作用的思想可以追溯到20世纪70年代末Schwyzer提出的“双价配体的消息-地址(message-address)”概念。该概念认为,一个双价配体由两个功能部分组成:一部分为“消息”(message),负责促进信号向效应系统的传导;另一部分为“地址”(address),通过增强配体与受体之间的特异性相互作用来提高选择性。在双位点配体中,地址部分通常由别构药效团承担,从而能够为那些正构位点高度保守的GPCR家族(例如毒蕈碱型乙酰胆碱受体或代谢型谷氨酸受体)提供亚型选择性。在这种情况下,别构药效团作为“地址”引导相对缺乏选择性的正构药效团“消息”。此外,由于双位点配体本身具有正构激动信号,其在内源性配体水平降低的神经退行性疾病治疗中也具有潜在优势。
然而,双位点配体的复杂性不仅体现在设计方面,也体现在其功能表征上。一些最初被认为是单价配体的化合物,后来被怀疑实际上属于双位点配体。来自多个研究团队的大量研究表明,许多曾被报道为别构激动剂的化合物(例如M1受体激动剂)实际上是双位点配体。这些配体能够结合于特定的别构位点并诱导具有功能选择性的M1受体激活,但在较高浓度下,它们还能够结合正构位点,并通常作为M2至M5受体的正构拮抗剂。同时,还存在一些分子开关能够消除其在别构位点上的结合能力,从而得到广谱正构毒蕈碱受体拮抗剂。
Conn研究团队的一项研究进一步揭示了M1别构(双位点)激动剂开发中的复杂性。在该研究中,两种结构相关的M1别构激动剂表现出受体储备依赖的药理特性,呈现出不同程度的部分激动效应,并在不同脑区诱导不同的生理反应,这与M1受体表达水平和受体储备差异有关,并与动物模型中的行为学效应相对应。此外,这些双位点配体还表现出配体偏向性信号传导:尽管在标准钙离子动员实验中表现出相似效应,但在β-arrestin募集以及ERK1/2磷酸化方面却表现出不同作用。这些结果表明,M1别构激动剂能够差异性地调控M1受体与不同信号通路之间的耦联关系,从而显著影响这些化合物在特定脑神经环路中的药理作用。因此,相比之下,通过增强内源性乙酰胆碱作用的M1 PAM可能是一种更为简单且可控的选择性M1激活策略。
随着GPCR别构调控研究的不断深入,双位点配体已经成为一个重要且具有发展潜力的研究方向。这类配体不仅可能成为新的分子探针和潜在治疗药物,其结构特性也为理解GPCR正构位点与别构位点之间的空间与热力学相互作用提供了重要线索。然而,目前仍缺乏一种完全明确的方法来鉴定双位点配体,这一问题可能需要依赖GPCR晶体结构解析或针对相关靶标的放射性配体技术的发展才能得到解决。
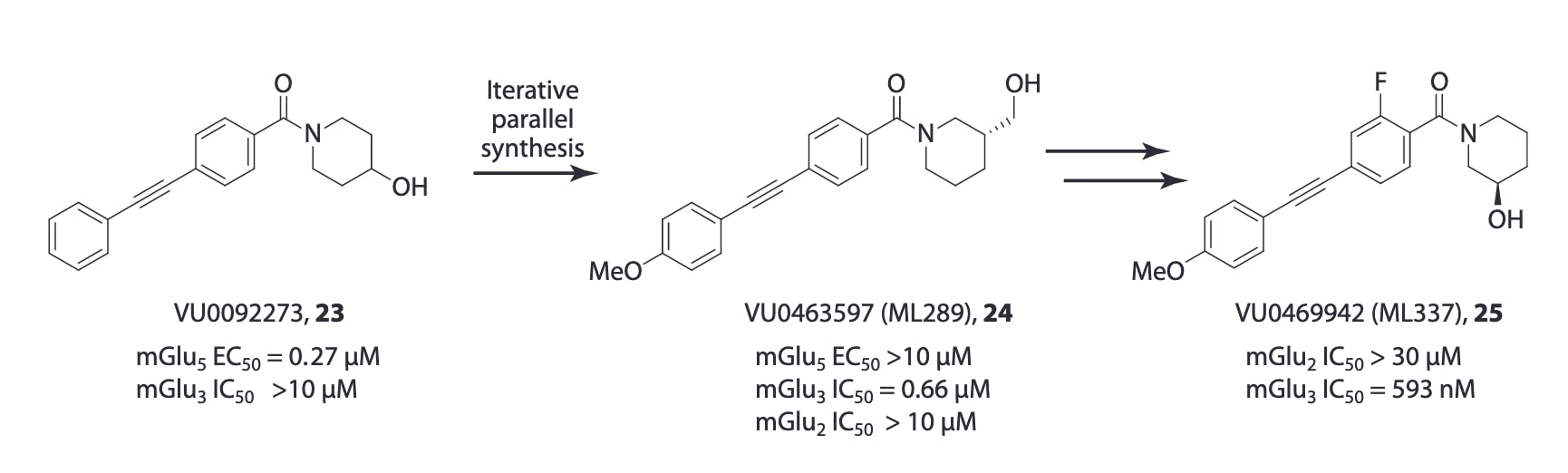
图7|利用分子开关获得新的亚型选择性别构调节剂。 从mGlu5 PAM VU0092273(23)出发,通过结构优化得到具有高选择性和高效力的mGlu3 NAM VU0469942(25)。
5 总结与结论
在过去十年中,对通过别构调控作用于多种分子靶标的理解以及其所带来的优势得到了显著提升。靶向GPCR的别构配体已经有部分药物成功上市,同时别构激酶抑制剂以及其他别构药物也正在针对多种中枢神经系统疾病和肿瘤适应症开展不同阶段的临床研究。在药物发现的早期阶段,别构配体凭借前所未有的选择性以及更优的理化性质,使研究者能够更深入地解析高度保守受体家族中不同成员的生物学功能及其潜在治疗价值。
尽管别构调控具有明显优势,但仍存在若干需要关注的问题,包括平缓的构效关系、化学或代谢产生的分子开关、配体偏向性信号传导在体内的生理影响以及不同物种之间可能存在的活性差异。然而,在过去五年中,针对别构位点设计和开发配体与药物的一系列关键原则和策略已经逐渐形成。这一研究方向为开发高度选择性的治疗药物提供了新的机遇,并有望在广泛的人类疾病领域中发挥重要作用。