Chem. Sci. 2021|蛋白质−蛋白质相互作用肽类抑制剂:引入构象约束后的生物物理、结构与细胞学影响
蛋白质−蛋白质相互作用(protein–protein interactions,PPIs)在细胞信号传导、转录调控和代谢网络中发挥核心作用,但由于其结合界面通常较大且结构相对平坦,使其长期被视为“难以成药”的靶点。近年来,利用肽类分子模拟天然蛋白界面关键结构单元成为调控PPIs的重要策略,其中以α螺旋为核心的肽类抑制剂尤为受到关注。然而,天然线性肽往往存在构象不稳定、易被蛋白酶降解以及细胞通透性较差等问题。通过在肽链中引入化学“订书”或其他构象约束,可以稳定其生物活性构象,从而在一定程度上提高结合亲和力、增强稳定性并改善细胞摄取能力。今天介绍的是发表于 Chemical Science 的一篇综述,系统总结了 构象受限肽在调控PPIs中的研究进展 ,重点讨论引入结构约束后对肽分子的生物物理性质、结构行为以及细胞水平表现所产生的影响,并为未来理性设计高效的PPI抑制剂提供重要参考。

Wang, H.; Dawber, R. S.; Zhang, P.; Walko, M.; Wilson, A. J.; Wang, X. Peptide-Based Inhibitors of Protein–Protein Interactions: Biophysical, Structural and Cellular Consequences of Introducing a Constraint. Chem. Sci. 2021, 12 (17), 5977–5993. https://doi.org/10.1039/D1SC00165E.
0 摘要
蛋白质-蛋白质相互作用(protein-protein interactions,PPIs)通过促进并调控单个蛋白质的功能,参与了绝大多数细胞过程。因此,PPIs被视为具有重要价值但同时极具挑战性的治疗干预靶点。近年来,构象受限肽的发展逐渐成为设计肽类PPI抑制剂的一种新兴策略,这类抑制剂通常以α螺旋结构为主要作用模式。通过在肽链中引入结构约束,可以带来多方面的潜在优势,例如提高结合亲和力、增强稳定性以及改善细胞穿透能力。 该策略的核心思想在于通过预组织化结构在结合前部分“支付”结合所需的熵成本,同时避免肽链转变为易被蛋白酶识别和降解的β链构象,并在一定程度上屏蔽亲水酰胺基团与疏水细胞膜之间的不利相互作用。
这一概念框架为基于经验设计肽类PPI抑制剂提供了清晰思路,被认为是推动PPI抑制剂药物发现的一条极具潜力且可能带来显著回报的路径。然而,在肽链中引入构象约束不仅会带来上述优势,还可能产生一系列更为微妙的影响,例如改变配体与受体之间的结合动力学、影响分子识别方式以及改变分子的理化性质。该综述系统总结了在不同约束化学策略和不同靶点背景下,引入结构约束对肽分子的生物物理性质、构象行为、结构特征以及细胞水平表现所产生的影响,同时强调了构象受限肽在该领域取得的显著成功,并讨论了未来设计中所面临的机遇与挑战。
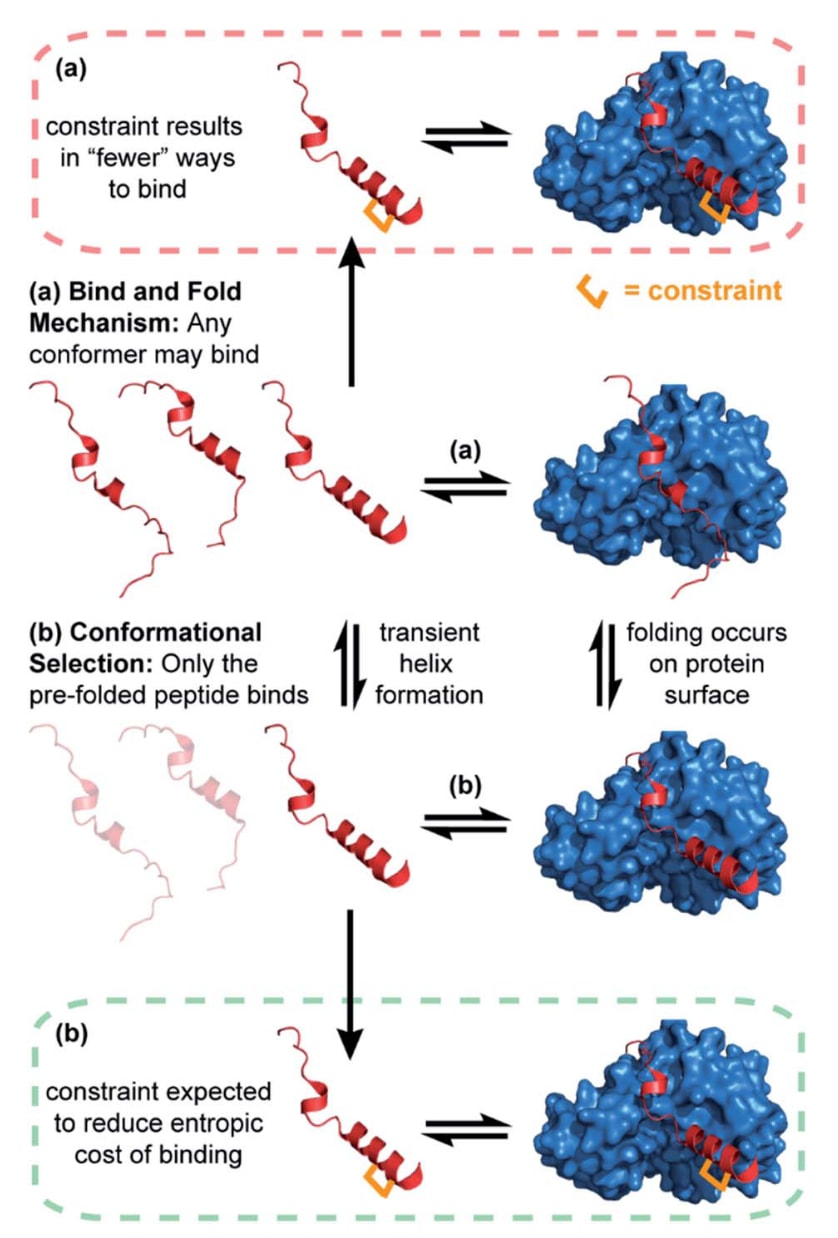
图1|不同肽-蛋白质相互作用结合机制示意图。 (a)“结合并折叠”(bind and fold)机制;(b)“构象选择”(conformational selection)机制。当结合依赖于特定构象时(b),引入构象约束通常可以提高结合亲和力(绿色虚线)。而在“结合并折叠”机制(a)中,引入构象约束可能对结合亲和力没有明显影响,甚至可能降低亲和力(红色虚线)。
1 引言
蛋白质-蛋白质相互作用(protein-protein interactions,PPIs)几乎参与了所有生物学过程,并与多种疾病密切相关。然而,在药物发现领域中,PPIs长期以来一直被认为是极具挑战性的竞争性(正构位点)抑制剂靶点,因为与传统药物靶点相比,这类相互作用通常涉及面积较大的蛋白质表面,并且结构特征相对较少。研究表明,α螺旋在人体PPI相互作用网络中具有较高的出现频率,因此被视为配体设计中的一种通用药效团。基于这一特征,由α螺旋介导的PPIs已成为开发选择性分子探针和药物候选物的重要研究方向,以满足众多尚未解决的治疗需求。
在这类PPI界面中,一个蛋白质的α螺旋通常嵌入结合伙伴蛋白表面的沟槽中。大量此类相互作用依赖于较短的肽序列基序,这些基序往往位于内在无序区域(intrinsically disordered regions,IDRs)之中,因此α螺旋结构通常是在PPI形成过程中被瞬时稳定下来的。通过晶体学和结构生物学分析,可以设计出在拓扑结构或空间排布上模拟α螺旋的肽分子或经过精心设计的小分子。 这类模拟物被称为“拟肽”(peptidomimetics),为开发PPI抑制剂提供了重要的起点,并已推动部分候选药物进入临床研究。
构象受限肽是拟肽分子中的一个重要分支,近年来逐渐成为调控PPIs的有力工具。通过化学方法在肽链中引入结构约束(通常称为“订书钉”或stapling),可以将肽稳定在其具有生物活性的α螺旋构象中,并带来多种优势,例如提高对蛋白酶的抗降解能力、增强在细胞环境中的稳定性、促进细胞摄取以及改善整体生物物理性质,相比天然序列往往表现出更优性能。
约束在序列中的位置会影响肽的螺旋含量,而所采用的化学连接方式同样会对其结构与功能产生影响。自Grubbs和Verdine率先发展烃链订书技术以来,研究领域已建立起丰富的化学工具来实现肽的构象约束。这些方法包括二硫键、内酰胺桥、氢键替代结构、烷撑链连接、基于巯基-烯反应形成的桥连、通过“点击化学”构建的三唑订书结构,以及多种超分子策略,其中一些方法还允许通过约束连接体进一步进行功能化修饰。面对如此多样的合成策略,一个自然的问题是:如何预测哪一种订书方式最适合特定体系。不同的化学连接体在同一肽序列中诱导α螺旋形成的能力并不相同,而当肽被限制在其生物活性的α螺旋构象时,最佳的柔性程度往往取决于具体靶标。
该综述概述了构象受限肽在生物物理性质和结构行为方面表现出的一些非经典特征,重点关注由α螺旋介导的PPIs,同时指出该策略已逐渐拓展至其他类型的PPI体系。文章汇集了近年来的一些新颖观察结果,强调不同订书类型在调控肽构象以及靶标结合方式方面可能产生的差异性影响。此外,还讨论了有关细胞穿透机制的最新研究进展,以及决定构象受限肽细胞摄取能力的重要结构特征。整体而言,这些内容为理性设计高效且具有良好细胞渗透能力的PPI抑制型构象受限肽提供了重要参考。
2 增强蛋白质结合亲和力
在其来源蛋白中以α螺旋形式存在的肽片段,在溶液中往往呈无序状态。当肽与蛋白质结合时,α螺旋结构通常通过“成核—延伸”的机制逐步形成。因此,螺旋折叠所带来的熵成本可能会削弱肽与靶蛋白之间的结合能力。通过引入化学连接体将肽限制在其具有生物活性的α螺旋构象中(即“订书”或stapling),理论上可以提前承担这部分熵成本,从而提高与靶蛋白的结合亲和力。然而,实际情况并非总是如此,这也表明在理解配体-受体相互作用时需要更加细致的理论框架。
2.1 结合动力学、热力学与结合机制的重要性
将肽预组织为其结合态的生物活性构象能够增强结合亲和力的观点,建立在若干假设之上。其中一个假设是靶蛋白仅在肽形成α螺旋结构时才进行识别。在这种通过“构象选择”(conformational selection)进行结合的情况下,对肽进行构象限制能够提高溶液中生物活性构象的平衡比例,从而使其更有效地与靶标发生相互作用。相反,如果靶蛋白能够识别未折叠的肽,则这一策略未必会带来同样的效果。当结构较为无序的肽在结合界面上被识别并随后发生折叠时,这一过程通常被称为“结合并折叠”(bind and fold)机制。在这种情况下,预组织化并受到约束的肽可能反而减少了可供结合的构象途径,从而导致结合和解离速率降低,并且对整体亲和力的影响可能很小甚至是负面的。
此外,预组织化对结合焓和结合熵的影响同样需要考虑。肽在折叠过程中,其焓变化来源于主链和侧链氢键的形成、电荷相互作用以及溶剂化状态变化等因素;同时,折叠的总熵变化则包括溶剂重新组织、分子的转动和平动自由度变化以及肽构象有序化等多方面贡献。对于溶液中的游离肽而言,未受约束的肽通常更加无序,水分子会围绕暴露的主链酰胺形成有序结构,而受约束的肽则较少出现这种情况。当肽折叠时,未受约束的肽能够在主链酰胺之间形成新的氢键,从而获得焓上的稳定贡献;而受约束的肽在这一方面的增益较小,但同时也较少受到水分子释放所带来的熵效应影响。由于焓和熵之间往往存在补偿效应(enthalpy-entropy compensation),折叠稳定性的净提升通常较为有限,折叠自由能变化ΔG往往仅相当于一次非共价相互作用的能量规模(通常小于5 kJ mol⁻¹)。因此,在不引入额外相互作用的情况下,预组织化所带来的稳定性提升往往有限。需要指出的是,这种框架仍然是一种简化描述,因为将受约束肽与天然序列直接进行能量比较并不完全合理;引入约束实际上主要提高的是未折叠状态的稳定性。
Miles等人在研究BH3家族肽的预组织化对BH3/BCL-2家族蛋白相互作用抑制能力的影响时,对BID和BIM的BH3结构域与MCL-1及BCL-xL的相互作用进行了分析。研究发现,引入烃链订书虽然提高了这些肽在溶液中生物活性α螺旋构象的比例,但并未增强其抑制活性,甚至部分受约束肽表现出明显的活性下降。共晶结构分析显示,约束的引入并未改变关键热点侧链的取向或肽在结合界面中的配准,也未与靶蛋白产生明显的空间冲突,因此活性下降无法通过静态结构解释。表面等离子共振(SPR)实验进一步表明,与天然序列相比,受约束肽的结合速率和解离速率均明显降低,这与“结合并折叠”的机制相一致。通过Van’t Hoff分析荧光各向异性结合实验数据发现,虽然受约束肽在结合时的熵成本确实降低,但这种有利的熵变化被不利的焓变化所抵消。
类似的焓-熵补偿现象也在组蛋白H3与ASF1相互作用的研究中被观察到。在该研究中,对H3肽(118–135位残基)在不同位置引入i,i+4烃链订书进行比较分析。结果显示,α螺旋形成程度不仅受到约束本身的影响,还显著依赖于约束在序列中的具体位置。这一现象并不令人意外,因为不同肽序列本身具有不同的α螺旋形成倾向,而引入非天然氨基酸构建约束结构会在不同程度上改变这一倾向。
在针对真核翻译起始因子4E(eIF4E)的研究中,也获得了关于肽约束效应的重要见解。eIF4E通过与竞争性结合伙伴4E-BP1和eIF4G相互作用来调控帽依赖型mRNA翻译,这两种蛋白均通过内在无序区域中的α螺旋基序与eIF4E的同一结合沟槽结合。分子动力学模拟表明,烃链订书的eIF4G肽在结合与未结合状态下均表现出不同的结构动力学行为。虽然订书可以稳定未结合状态下的螺旋构象,但也可能偏向某些亚稳态构象,这些构象在结合时仍需要发生重排才能使关键侧链达到正确的识别取向,从而阻碍结合过程。实验和结构研究进一步证实,稳定与结合态一致的溶液构象对于提高结合能力至关重要。基于这一认识,通过理性设计获得了能够在eIF4E表面未被利用区域形成相互作用、并具有更长靶标驻留时间的订书肽。
Gallagher等人的研究也得出了类似结论。研究首先比较了线性4E-BP1和eIF4G肽在溶液中的螺旋倾向,发现4E-BP1本身具有更高的螺旋含量。SPR实验进一步分析了温度和盐浓度对结合行为的影响,结果表明eIF4G的结合依赖于电荷相互作用并且结合动力学随温度变化,而4E-BP1则相对稳定。尽管两者在结合态结构上相似,但其结合机制并不相同:4E-BP1对eIF4E具有更高的互补性,并形成更加稳定的复合物。进一步研究发现,当对4E-BP1进行烃链订书时,其亲和力和生物活性均得到提升,说明该体系主要遵循构象选择机制;而对eIF4G进行订书则形成了非螺旋的大环肽,其亲和力反而低于线性序列。这些结果再次强调,在决定是否引入构象约束之前,理解无序肽的结合机制和动力学行为至关重要。
在Aurora-A有丝分裂激酶与微管相关蛋白TPX2之间的α螺旋介导PPI研究中,Jamieson等人采用烃链订书策略设计TPX2肽。结果表明,该订书肽不仅具有更高的结合亲和力,还能够模拟TPX2激活Aurora-A自磷酸化的功能。然而,等温滴定量热(ITC)实验显示,尽管约束提高了螺旋比例并增强了亲和力,其结合熵却比天然肽更加不利,而结合焓则更加有利。晶体结构分析解释了这一现象:烃链订书本身并未直接与Aurora-A表面接触,关键热点残基的空间取向也基本一致,但订书使螺旋延长了一个额外的螺旋圈,从而改变了结合构象,使Glu36和Lys38两个带电侧链能够与Aurora-A形成额外的氢键相互作用。因此,不利的熵变化可以归因于形成更长、更有序的螺旋结构,而这些变化同时带来了额外的有利焓贡献。
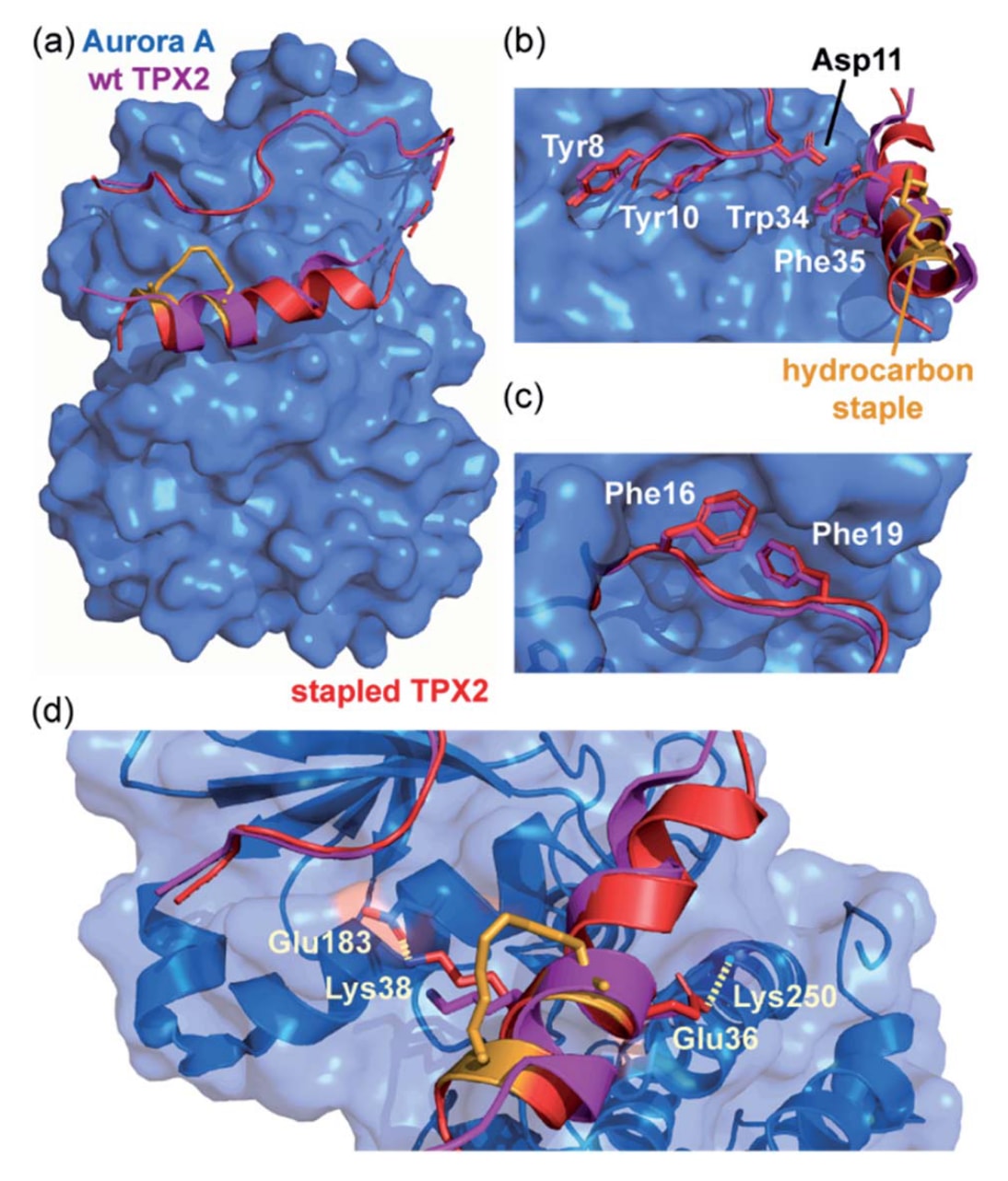
图2|未受约束与受约束的Aurora-A结合TPX2肽的比较。 (a)Aurora-A(蓝色)与订书TPX2肽(红色,烃链订书:橙色,PDB:5LXM)复合物结构,并叠加天然TPX2(紫色,PDB:1OL5)。为突出订书TPX2与天然TPX2之间保守的结合模式,将已知对Aurora-A结合至关重要的TPX2残基以棒状形式显示;(b)Tyr8、Tyr10、Asp11、Trp34和Phe35;(c)Phe16和Phe19;(d)Lys38和Glu36的取向发生变化,从而促进肽与Aurora-A之间额外的静电相互作用(虚线)。
在针对HIF-1α/p300相互作用的研究中,也观察到了类似现象。该PPI参与细胞氧水平调控,并常被癌细胞利用以支持肿瘤生长。研究通过i,i+4位半胱氨酸与二溴马来酰亚胺反应形成约束结构。结果显示,其中一种二溴马来酰亚胺订书肽对p300的结合亲和力明显高于未受约束的天然肽。然而,圆二色谱实验表明这种亲和力提升并未伴随α螺旋含量的增加。分子动力学模拟显示,无论受约束还是未受约束的肽,在与p300结合时螺旋特征都会增强,而订书肽在结合态中表现出更显著的稳定化。因此,亲和力提升被认为源于订书结构稳定了结合态构象,这一结论也得到了CD差谱实验的支持。
类似地,在Exoenzyme S来源肽与14-3-3蛋白的相互作用研究中,也发现引入约束后结合态行为的变化对相互作用具有重要影响。在该体系中,亲和力的提升被认为与配体-受体复合物中肽段动力学增强有关。
Strizhak等人的研究中构建了一系列基于噬菌体展示技术发现的p53/MDM2相互作用肽的类似物。这些肽在
通过基于色氨酸荧光淬灭的竞争结合实验发现了一个有趣现象:带有“开环”DAE光异构体的受限肽与MDM2的结合能力始终强于其“闭环”对应物。为了进一步理解结合的热力学特征,研究利用等温滴定量热(ITC)进行了测定。结果显示,“闭环”异构体的结合主要由焓驱动,而“开环”异构体则表现出更显著的熵贡献。
随后对亲和力最高的类似物(
尽管“开环”异构体具有更大的构象自由度,但结构数据表明其能够形成额外的非共价相互作用。然而,与“闭环”形式相比,其结合过程中为何表现出更大的熵贡献,目前仍未完全解释。尽管如此,该研究清楚地表明,在构象受限肽中,连接体本身也可能直接参与分子识别并影响结合行为,这一现象将在后续部分进一步讨论。
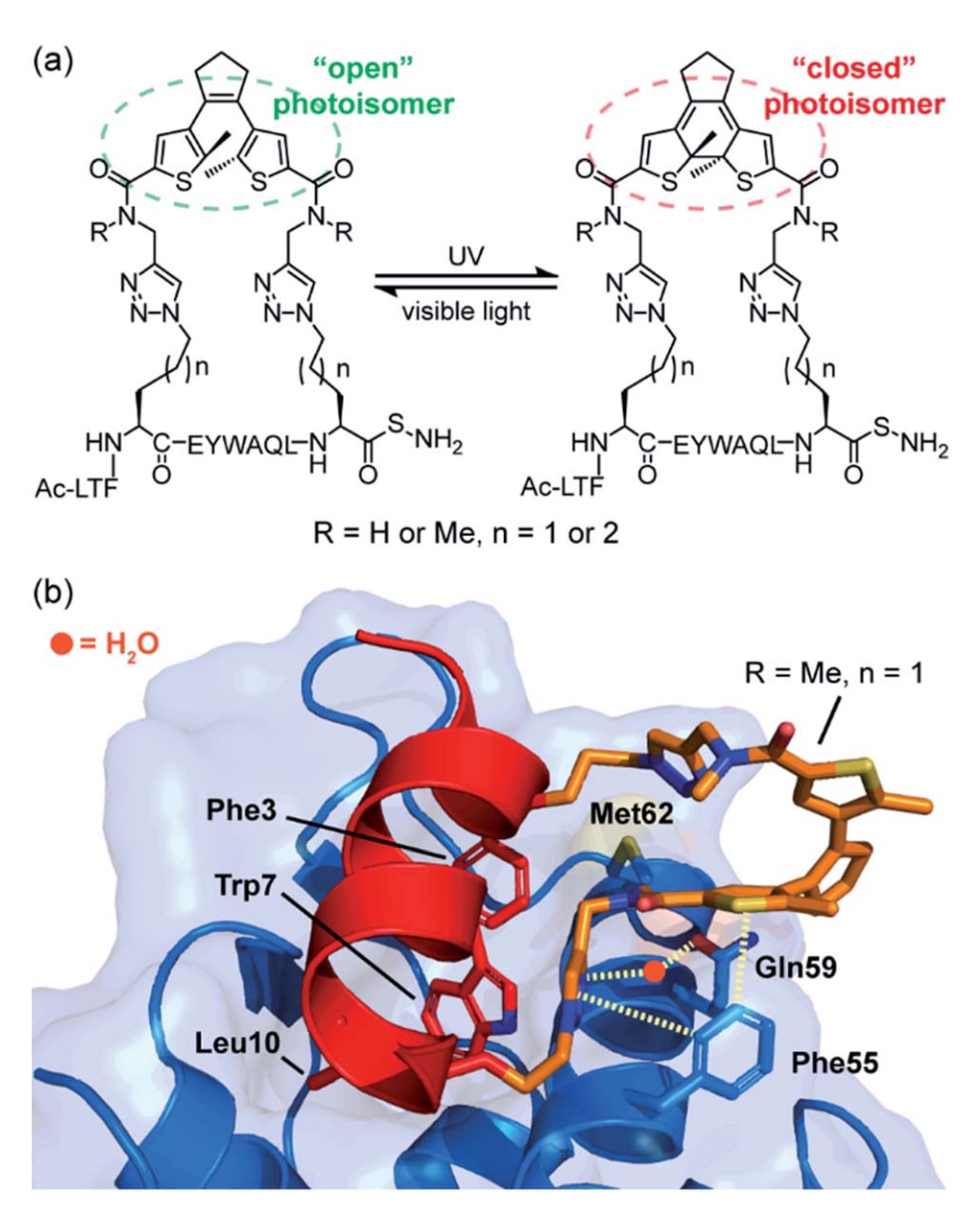
图3|具有可光切换约束结构并表现出不同热力学特征的MDM2结合肽。 (a)用于靶向p53/MDM2相互作用的DAE订书光异构体结构,包括“开环”和“闭环”两种形式(
2.2 形成更多相互作用:订书结构与靶标之间的直接作用
在设计用于调控α螺旋介导PPI的构象受限肽时,约束结构通常被引入到α螺旋的溶剂暴露表面,以避免与蛋白质结合界面产生空间冲突,或干扰关键热点残基的相互作用。然而,在一些研究中发现,化学连接体本身也可以与靶蛋白形成有利的相互作用,从而增强结合能力,甚至改变原有的结合方式。如果能够在设计中使约束结构直接与靶蛋白形成额外的有利接触,可能为构象受限肽的优化提供新的策略。以下实例主要基于共晶结构分析,展示了这种现象,并揭示了在有意设计此类约束时所面临的一些挑战。
Walensky团队筛选了一系列稳定化α螺旋BCL-2结构域(stabilized alpha-helix of BCL-2 domains,SAHBs),并发现MCL-1蛋白自身的BH3螺旋序列可以作为MCL-1的抑制剂。通过定点突变和订书位置扫描(staple scanning)策略,研究进一步获得了一种优化的烃链订书MCL-1肽(MCL-1 SAHBD)。为了阐明关键相互作用,解析了该订书肽与MCL-1复合物的晶体结构(图4a)。结构分析显示,烃链订书本身能够与MCL-1结合位点边缘形成明确的疏水相互作用(图4a)。此外,所引入的二取代非天然氨基酸中的一个甲基基团占据了由Gly262
Phillips等人的研究则通过针对核受体(nuclear receptors,NRs)开发订书肽抑制剂,进一步揭示了设计构象受限肽时需要考虑的重要因素。研究设计了一种11肽长的订书肽Ac-H-S5-ILH-S5-LLQDS-NH2(SP1,其中S5表示(S)-2-(4-pentenyl)alanine),用于靶向雌激素受体(estrogen receptor,ER)的共激活因子结合位点,并通过订书位置扫描确定最优的约束位置。
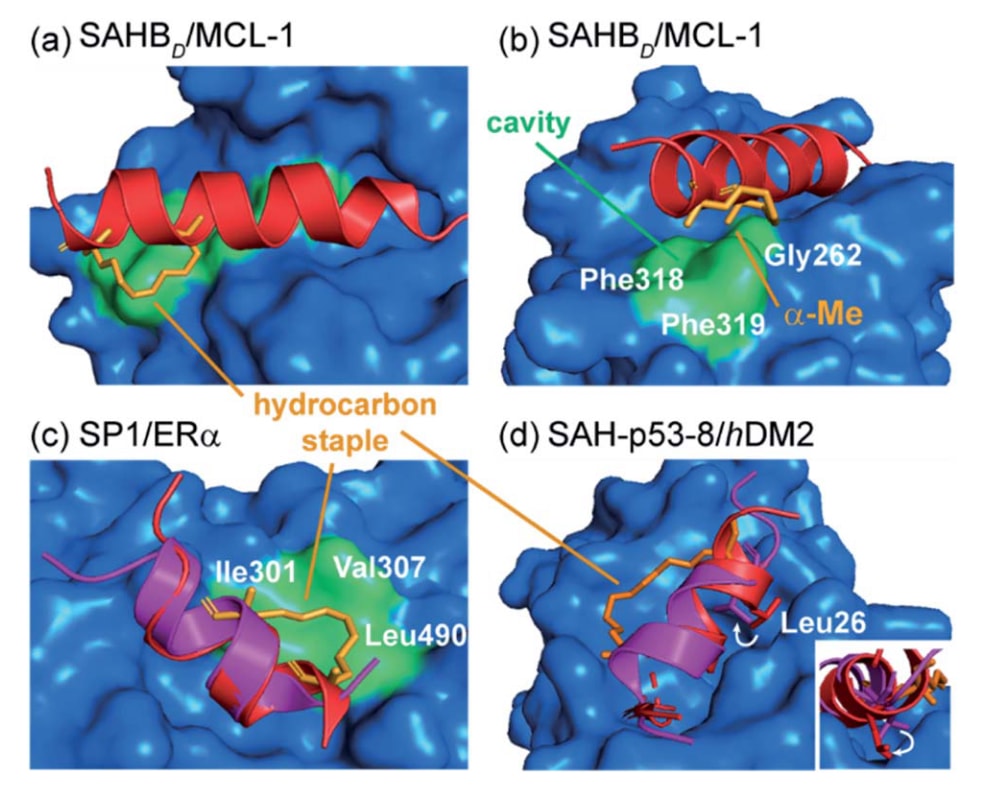
图4|蛋白质结合的构象受限肽共晶结构,展示约束结构与蛋白质之间可能形成的相互作用。 (a)MCL-1 SAHBD/MCL-1复合物的晶体结构(PDB:3MK8),其中MCL-1 SAHBD的烃链订书结构(橙色)在核心结合位点边缘形成额外的疏水相互作用;(b)α,α-二取代基团中的一个甲基占据由MCL-1的Gly262、Phe318和Phe319(绿色)形成的沟槽;(c)雌激素受体ERα结合结构中天然序列(紫色,PDB:2QGT)与订书肽SP1(红色,PDB:2YJD)的比较;(d)hDM2结合结构中天然p53肽(紫色,PDB:1YCR)与订书肽(红色,PDB:3V3B)的比较。
对这些复合物结构以及游离肽构象的研究表明,约束结构不仅会稳定肽的构象,还可能直接参与与靶蛋白的相互作用。对于活性最强的订书肽SP1,其与ERα配体结合结构域形成复合物时,圆二色谱(CD)和核磁共振(NMR)分析显示,引入烃链订书显著提高了肽的α螺旋含量。然而,SP1/ERα复合物的晶体结构进一步揭示,疏水性的订书结构本身能够与ERα表面由Val307、Ile310和Leu490形成的疏水区域产生有利接触(图4c)。与共激活因子蛋白-肽复合物结构(2QGT)比较发现,该订书肽的螺旋发生了约四分之一圈的偏移,使原本参与结合的残基位置整体错位一个氨基酸。因此,烃链订书不仅限制了肽的构象,还诱导了一种非经典的结合模式,这种变化可能带来潜在的非特异性效应。
随后,Baek等人报道了烃链订书p53肽SAH-p53-8与其结合伙伴hDM2的复合物晶体结构。结构显示,订书结构占据了hDM2中p53结合位点边缘的一片疏水区域,约贡献了肽与蛋白之间总接触面积的10%,这很可能增强了整体结合亲和力。与此同时,订书使受约束肽中Leu26
类似现象也出现在ATSP-7041的研究中。ATSP-7041是一种由Aileron公司开发的临床阶段肽类抑制剂,可同时靶向hDM2和hDMX。Ghadessy团队进一步报道了烃链订书肽M06与hDM2的复合物晶体结构,M06是Brown等人此前报道序列的一个变体。结构分析表明,该订书结构能够与hDM2的疏水残基形成有利的堆积相互作用。此外,在M06/hDM2复合物结构中,肽结合沟槽在靠近肽C端的位置被hDM2的螺旋“铰链”区域覆盖(图5a)。
将该结构与Baek等人报道的SAH-p53-8/hDM2复合物进行比较可以发现显著差异(图5b)。在M06复合物中,由于M06形成的螺旋较短,无法像SAH-p53-8那样完全填满hDM2的结合口袋,因此hDM2的铰链区域会形成螺旋结构并覆盖该口袋,从而实现对M06更紧密的包裹(图5c)。而在SAH-p53-8/hDM2结构中,hDM2的Tyr100向口袋内部延伸,并通过与Asn29
这些研究表明,尽管约束结构能够直接与靶蛋白形成相互作用,但这种相互作用往往伴随着蛋白或肽构象的变化。因此,在设计构象约束以优化与靶蛋白的非共价相互作用时,需要同时考虑这些潜在的结构重排,这也增加了构象受限肽理性设计的复杂性。
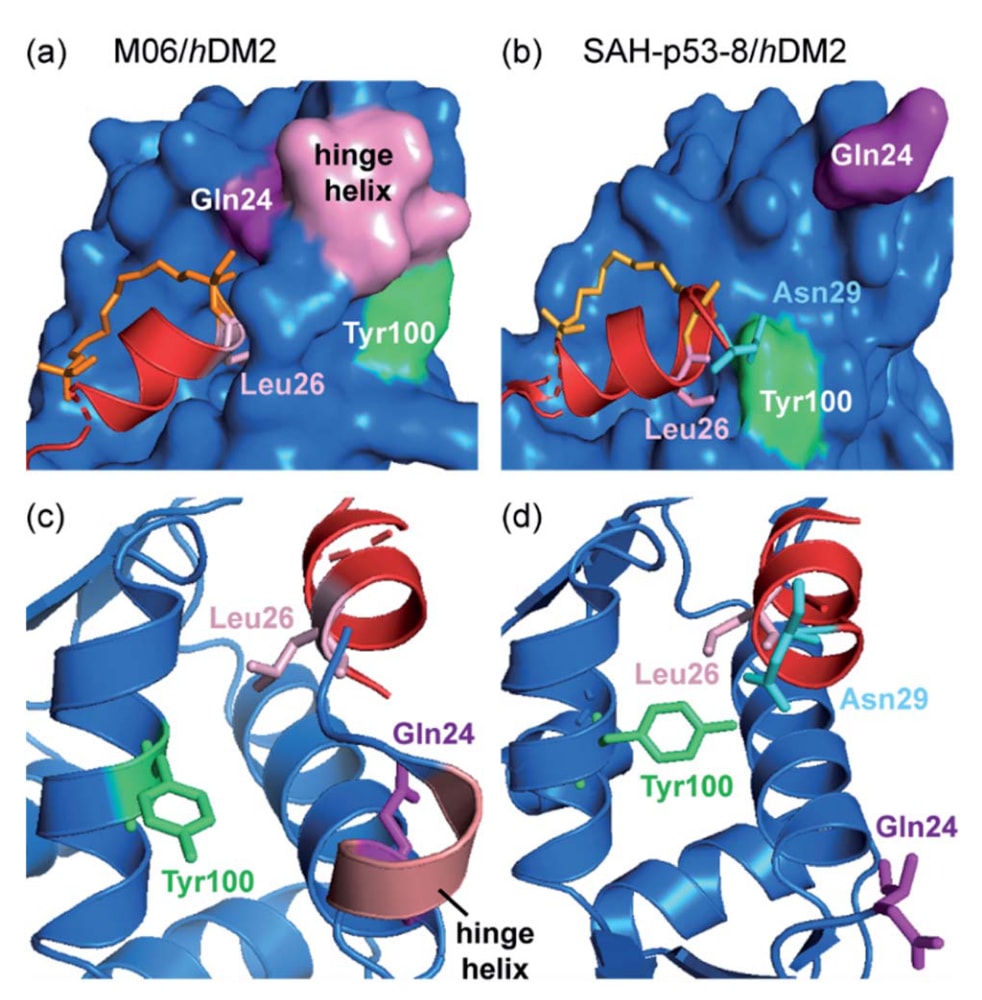
图5|构象受限p53/hDM2复合物结构比较。 (a)在M06复合物的晶体结构中形成的结合位点被hDM2“盖子”(lid)区域的“铰链”螺旋封闭(PDB:4UMN);(b)hDM2与SAH-8结合时形成的结合口袋结构(PDB:3V3B);(c)“铰链”螺旋覆盖在结合口袋底部;(d)Tyr100(绿色)处于“闭合”构象,并与Asn29形成氢键相互作用。
2.3 订书结构:对肽构象与靶标结合的影响
如前文所述,用于限制肽构象的化学订书结构在某些情况下能够直接与靶蛋白发生相互作用,从而增强结合亲和力。因此,对约束结构进行细微调整,例如α位的立体化学、α位取代基类型、烃链的额外支化(例如γ位甲基)、订书插入位置以及连接体长度,都可能显著影响构象受限肽的行为。在烃链订书技术的发展过程中,人们已经注意到,仅仅改变连接体长度的微小差异,就可能对订书反应效率以及肽结构产生明显影响。下面的研究实例展示了订书结构在调控肽构象和靶标结合方面的多样作用。
传统的烃链订书通常通过在
Grossmann团队随后在研究靶向三聚体核转录因子Y(NF-Y)复合物的构象受限肽时,发现α-甲基化可能对结合行为产生显著影响。研究最初将
Grossmann团队还研究了改变双取代氨基酸α位较小烷基取代基的影响。此前研究报道了一种来源于致病蛋白ExoS的烃链交联大环肽,用于靶向人类适配蛋白14–3–3的结合界面。该肽最初由11个关键氨基酸组成,并在第3和第6位引入R和S构型的α-甲基、α-烯基氨基酸(X(Me)R3和X(Me)S6),通过连接形成八元烃链桥,并在与14–3–3结合时呈现不规则结构。这一研究表明,订书策略不仅可以稳定α螺旋,还可用于稳定其他识别基序。随后通过截短实验确定保持结合能力的最小序列,并系统地将X(Me)R3和X(Me)S6中的α-甲基替换为氢或更疏水的乙基,共得到七种变体。结果显示,含至少一个氢取代基的变体均出现亲和力下降,而引入α-乙基的变体则表现出更强的结合能力。特别是在第3位引入乙基时亲和力提升明显,而在第6位引入乙基则影响较小,因此两个位置的乙基效应并不呈简单叠加。分子动力学模拟表明,游离肽在溶液中主要存在两类优势构象,而烷基取代基能够改变构象分布,使其偏向于亲和力更高的构象。同时,与logD值的相关性也表明疏水性因素可能发挥作用。随后解析的优化大环肽与14–3–3复合物晶体结构进一步显示,第3位的α-乙基能够更深入地插入蛋白表面的疏水腔体(cavity 1),相比之下较小的α-甲基则难以实现这种作用。因此,将α-甲基替换为α-乙基这一微小结构变化能够同时稳定游离态和结合态构象,从而提高结合能力。
此外,还开发了具有不同连接构型和长度的构象受限肽(例如
为了模拟亮氨酸和异亮氨酸等支链疏水侧链的作用,Speltz等人设计了一类在订书氨基酸S5的γ位引入甲基的非天然氨基酸(
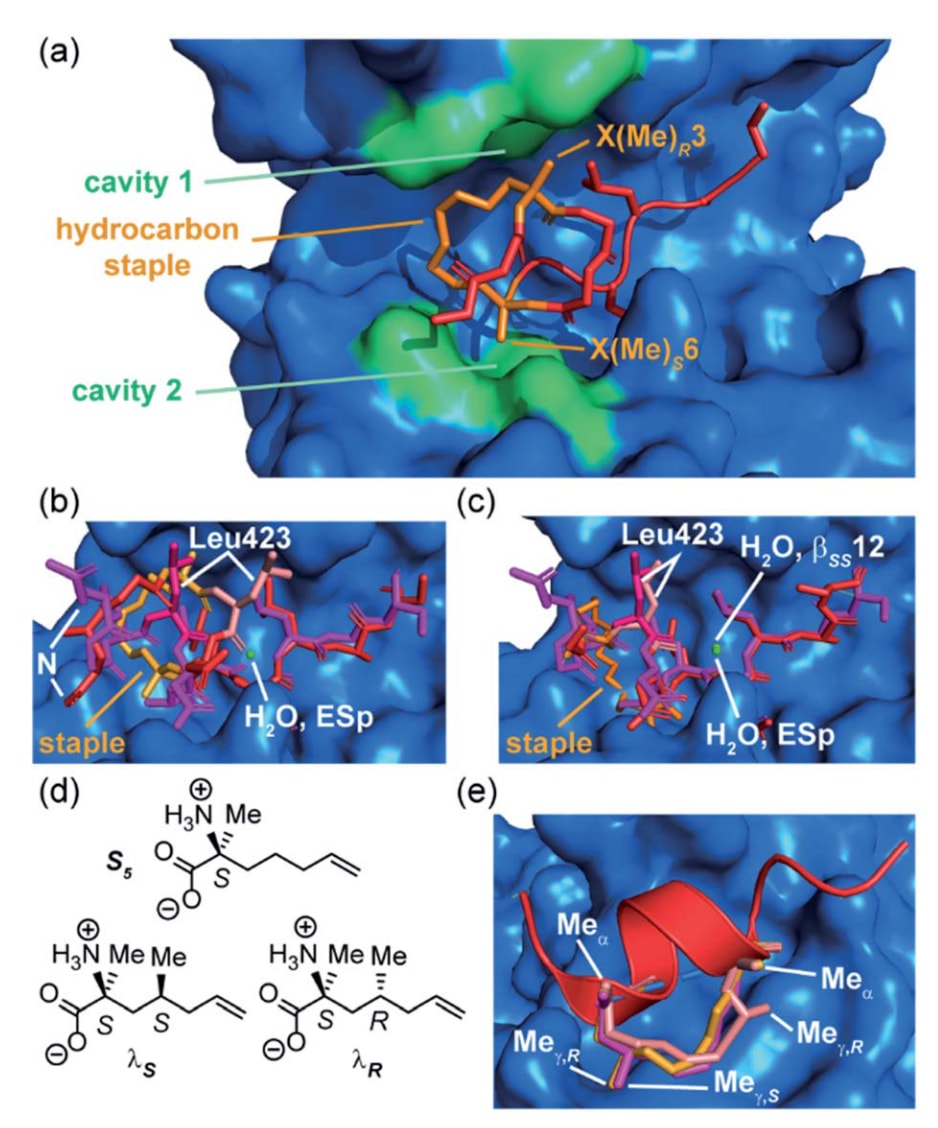
图6|订书结构支化对其行为的影响。 (a)烃链交联大环肽(红色,烃链订书:橙色,PDB:4N7Y)与14–3–3蛋白(蓝色)复合物的晶体结构。对14–3–3蛋白表面的计算分析显示,在氨基酸X(Me)R3和X(Me)S6附近存在两个空腔(cavity 1和2,绿色),可能作为优化结合亲和力的潜在位点;(b)ESp(紫色)与bRS8(红色,PDB:4N7G)结合14–3–3时的晶体结构比较;(c)ESp(紫色,PDB:4N7G)与bSS12(红色,PDB:4N84)结合14–3–3时的晶体结构比较;(d)支化订书氨基酸S5、lS和lR的结构;(e)SRC2-SP1(浅红色,PDB:5DXB)、SRC2-SP2(橙色,PDB:5HYR)和SRC2-SP3(紫色,PDB:5DX3)通过调整构象以缓解α-甲基与γ-甲基之间的syn-pentane相互作用。
2.4 约束类型的影响
Spring团队的研究表明,约束结构与靶蛋白形成有利相互作用的能力并不限于烃链订书。研究以来源于噬菌体筛选的PMI/PDI肽为基础,通过一种应变促进的炔-叠氮环加成反应(strain-promoted alkyne–azide cycloaddition,SPAAC)构建MDM2结合肽。这种订书策略被用于加速具有细胞活性的订书肽的发现。通过原位订书与筛选相结合的方法,研究鉴定出一种具有更高蛋白酶稳定性并且对MDM2具有纳摩尔级结合能力的订书肽。随后解析的复合物晶体结构证实,该肽在结合时形成α螺旋构象,并且订书结构中双三唑(bis(triazolyl))连接体呈现反向区域连接方式。结构分析显示,该约束肽能够正确排列关键的结合三联体残基Phe3
另一种用于稳定肽α螺旋构象的策略是氢键替代结构(hydrogen bond surrogate,HBS)。这种方法能够在一定程度上避免约束结构与蛋白表面产生不利相互作用。Douse等人比较了烃链订书与HBS策略在靶向恶性疟原虫(Plasmodium falciparum)肌球蛋白A(myosin A,myoA)与myoA尾部相互作用蛋白(MTIP)相互作用中的表现。研究发现,采用HBS约束的myoA肽与天然myoA肽表现出相似的抑制活性,这与其设计策略一致,因为HBS约束被刻意放置在myoA尾部螺旋的N端区域,位于拥挤的MTIP结合位点之外(图8a)。然而,当在同一体系中引入烃链订书时,肽的抑制活性反而下降。
通过比较这些受限肽与野生型肽的晶体结构可以解释这一现象。首先,MTIP的整体折叠结构保持不变,其两个结构域仍然夹持在受限myoA肽两侧,而烃链订书的空间走向与被替换的疏水残基基本一致(图8a)。由于HBS约束位于螺旋N端,因此其连接体不会与拥挤的MTIP结合沟槽产生空间冲突。相反,烃链订书位于螺旋中部,其脂肪链需要插入MTIP C端结构域中的一个较小疏水核心区域,从而阻碍了靶蛋白与受限肽之间形成最优相互作用(图8b)。
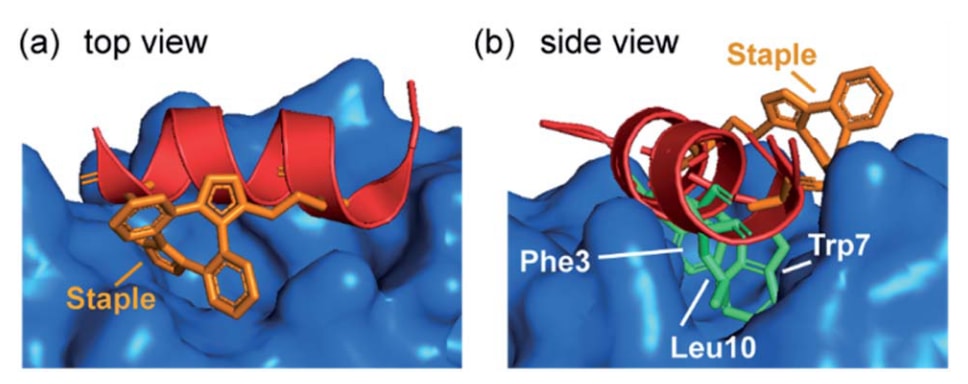
图7|双三唑(bis(triazolyl))订书肽与MDM2复合物的晶体结构(PDB:5AFG),展示了该肽的α螺旋构象以及订书结构的反向区域连接方式。
3 构象受限肽细胞摄取的决定因素
将构象受限肽用于调控细胞内蛋白质-蛋白质相互作用(PPIs)的一个潜在优势是其可能具有更好的细胞摄取能力。Walensky团队在2004年的研究中报道,一种烃链订书的α螺旋BID BH3肽不仅具有良好的细胞通透性,还能够在小鼠人类白血病异种移植模型中表现出体内抑制活性。这一发现推动了订书策略在其他细胞内靶点中的广泛应用,例如BCL-2家族相互作用以及p53/hDM2相互作用。尽管多种订书肽在细胞内PPI抑制方面表现出潜在活性,但仍需谨慎区分这些效应是否真正来源于靶点作用机制,并明确其与细胞通透性的关系。
Czabotar等人研究了一种来源于BIM BH3序列的烃链订书肽BimSAHB,发现其对BCL-2、BCL-xL、BCL-w和MCL-1的结合亲和力均低于未受约束的对应肽。此外,在小鼠胚胎成纤维细胞(MEF)和Jurkat细胞中并未观察到BimSAHB诱导的凋亡,而在细胞裂解液中则可以观察到活性。这表明该订书肽在细胞模型中活性降低可能同时源于结合亲和力下降以及细胞通透性不足。值得注意的是,该序列与Walensky团队报道的另一种订书肽BIM SAHBA1不同,后者在活细胞中表现出显著细胞毒性并能够激活caspase 3/7。进一步研究表明,BIM SAHBA1主要通过内体途径进入细胞,而非通过破坏细胞膜进入,并且主要定位于线粒体和多囊泡体。Walensky团队还指出另一序列BimSAHB(BIM SAHBA2)具有负电荷净值,这可能导致细胞通透性较差,这一结论与此前研究一致:在一条21肽序列中仅将一个Arg替换为Asp就会显著降低细胞摄取能力。这些结果说明,即使是细微的序列变化也可能显著改变细胞通透性。
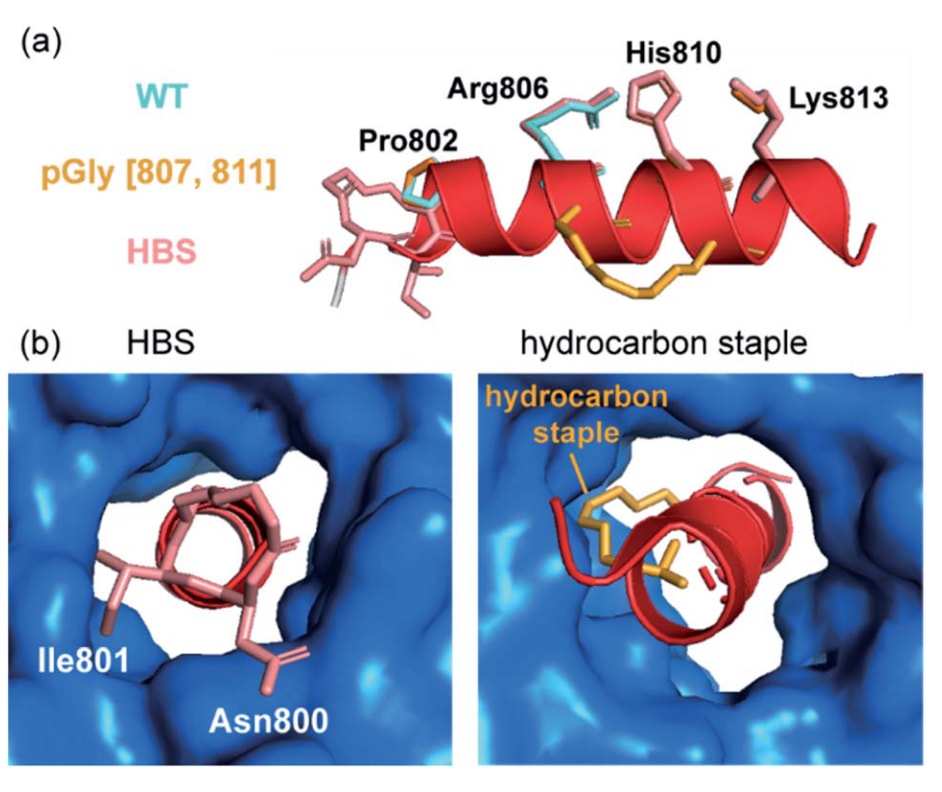
图8|构象受限肽作为MyoA/MTIP相互作用抑制剂。 (a)不同肽在与MTIP(蓝色)形成复合物时形成的螺旋结构比较:天然myoA尾部肽(侧链为青绿色,PDB:4AOM)、烃链订书肽(侧链为橙色,PDB:4MZK)以及HBS约束的myoA肽(鲑红色,PDB:4MZL);(b)HBS结构位于myoA肽螺旋的N端,而烃链订书位于螺旋中央位置。
尽管对某些订书肽的细胞进入机制已有较多研究,但仍存在许多未解问题。一些研究发现,构象受限肽可能通过破坏细胞膜导致细胞裂解,这种机制会引发非特异性毒性,因此并不理想。目前普遍认为构象受限肽进入细胞的主要途径是ATP依赖的内吞作用。细胞穿透肽(cell-penetrating peptides,CPPs)由于具有天然细胞通透性而被广泛用于细胞研究。Verdine团队比较了三种野生型CPP及其烃链订书版本的细胞进入机制,结果发现订书CPP并非完全依赖ATP驱动的途径,而是涉及多种机制,其中ATP非依赖型内吞占主导。然而,构象受限肽从内体逃逸进入细胞质的过程仍然研究较少。除内吞外,CPP研究中还观察到其他ATP非依赖机制,例如孔道形成和细胞膜破坏等,这些机制同样可能适用于订书肽。例如Fairlie团队报道,将乳内酰胺桥替换为烃链订书后会导致细胞裂解。
Li团队开发了一种重组酶增强双分子荧光素酶互补平台(ReBiL),用于在活细胞中检测弱PPI,并用于研究订书肽行为。在该体系中测试了三种对hDM2或hDMX具有较高体外亲和力的烃链订书肽SAHp53-8、sMTide-02和ATSP-7041。尽管这些肽在体外结合实验中优于小分子抑制剂Nutlin-3a,但在活细胞中几乎没有抑制活性。在无血清条件下,订书肽在细胞中的活性增强并伴随细胞膜破坏,而血清对细胞裂解液中的相互作用没有影响,这表明血清蛋白可能通过抑制膜破坏而降低肽进入细胞的能力。
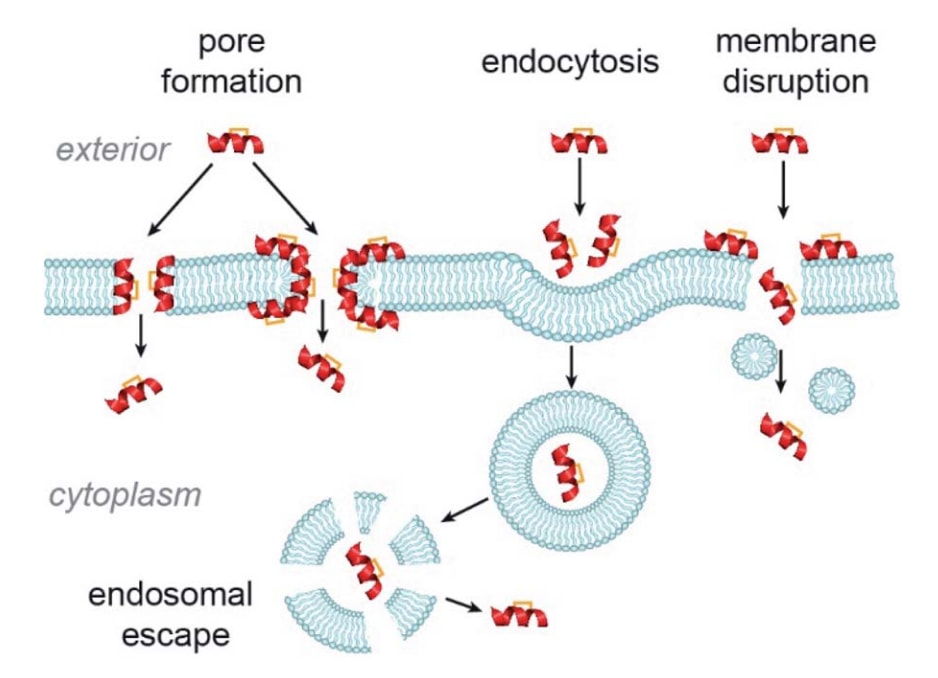
图9|构象受限肽可能的细胞内化机制示意图。
近年来,通过进一步修饰构象受限肽来提高细胞通透性也被证明是一种有效策略,例如引入细胞穿透基序或促进自组装的片段。然而,额外功能的引入可能带来新的空间干扰或分子识别效应,同时增加合成复杂度并引入新的理化性质问题。因此,不依赖额外修饰的构象受限肽在药物开发中通常更具吸引力,但其设计目前仍然主要依赖经验。
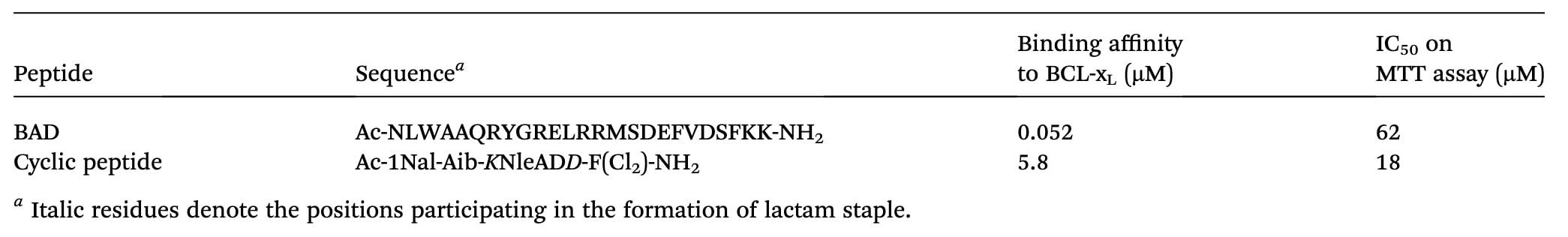
为了实现更理性的设计,需要理解影响细胞通透性的关键因素。Lin团队发现,一种针对MCL-1 BH3序列设计的构象受限肽,其细胞摄取能力与HPLC保留时间高度相关,说明疏水性较高的肽通常具有更好的细胞通透性。随后研究表明,对订书肽中的Ala进行N-甲基化可进一步提高细胞摄取能力,这表明通过屏蔽极性基团可以促进细胞进入,而N-甲基化在天然产物和环肽的细胞通透性中同样被认为具有重要作用。Fairlie团队也发现,引入疏水氨基酸残基的点突变能够提高细胞实验中的活性。例如在BAD BH3序列中引入Norleucine、1-萘丙氨酸、氨基异丁酸以及3,4-二氯苯丙氨酸等疏水残基后,虽然体外结合亲和力降低,但细胞活性提高,说明增强细胞摄取可能是主要原因。
表1|BIM SAHBA1与BIM SAHBA2的肽序列、生物物理性质、细胞效应以及细胞通透性的比较。

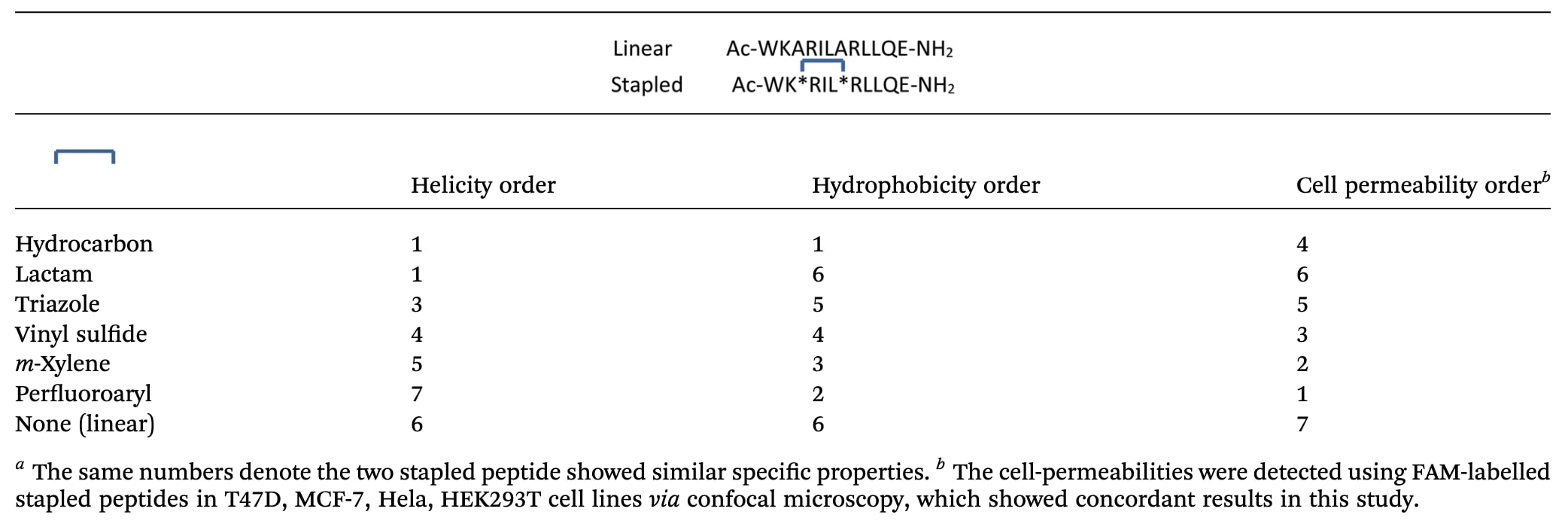
除序列疏水性外,连接体本身的疏水性也会影响细胞通透性。Li团队比较了多种不同约束类型的肽,包括烃链订书、乳内酰胺桥、三唑、乙烯硫醚、间二甲苯以及全氟芳基连接体。结果显示,具有更疏水连接体(例如烃链和全氟芳基)的订书肽在多种细胞系中的通透性比线性肽或亲水连接体肽高2–10倍,并且细胞通透性与反相色谱和流式细胞术测得的疏水性指标高度相关。
肽的α螺旋含量同样被认为会影响细胞摄取。Verdine团队研究了烃链订书中非天然氨基酸绝对构型对α螺旋形成和细胞摄取的影响,发现将(S,S)构型替换为(R,R)构型会降低螺旋含量并减少细胞进入。Li团队进一步通过在订书连接体中引入不同取代基(氢、甲基、乙基、异丙基、苯基和苄基)来构建手性中心,发现这些修饰会改变α螺旋含量,并且细胞通透性总体上与螺旋含量相关,但也存在例外。Ulrich团队的研究也指出,当肽与脂质膜结合时形成的α螺旋含量比溶液中的螺旋含量更能预测细胞摄取能力。不过Futaki团队的研究则认为疏水性比螺旋含量更重要,在一系列靶向p53/DM2的肽中,疏水性较高的未订书肽反而表现出更高的细胞摄取能力。此外,也报道了非螺旋结构但具有细胞通透性的订书肽,例如基于核定位信号(NLS)序列的双三唑订书肽。
表2|野生型BAD肽与环状肽在序列、结合亲和力以及MTT实验活性方面的比较。
净电荷也是影响细胞通透性的关键因素。Verdine团队发现,当肽的净电荷从+1增加到+5时,无论是单订书还是“缝合”(stitched)双订书肽,其细胞摄取能力都会提高,但当净电荷超过+7时,通透性反而下降。总体来看,构象受限肽的细胞摄取受到多种因素共同影响,包括疏水性、α螺旋含量、净电荷以及血清环境等。Walensky团队通过高通量荧光显微技术系统研究了这些因素对细胞摄取的影响,发现总体疏水性、净电荷、α螺旋含量以及订书位置都是关键决定因素。其中,适度而非过高的疏水性有利于细胞通透性;同时,将订书从亲水面移动到疏水面可以扩大疏水表面并提高细胞摄取能力。此外,较高的α螺旋含量(约61–86%)以及适当的等电点也有利于细胞进入,但过高疏水性结合较低pI值则容易引起细胞裂解。
Fairlie团队利用流式细胞术定量分析了一系列FITC标记订书肽在无血清条件下的细胞通透性,发现细胞摄取与连续疏水表面积(cHSA)以及疏水矩(mH)相关。缺乏两亲结构的肽无法进入细胞,而缺乏正电荷的两亲肽也表现出较差通透性。具有连续疏水表面和带电表面的肽则表现出明显更高的细胞进入能力。此外,该研究还发现烃链订书比乳内酰胺桥更容易引起细胞裂解。
表3|靶向ER共激活因子的模型肽在α螺旋含量、疏水性以及细胞通透性方面的比较。
最后,虽然由全D型氨基酸组成的肽通常具有较高的抗蛋白酶稳定性,但其细胞通透性往往较差。近期Kannan等人开发了一系列烃链订书和“缝合”结构的全D型氨基酸肽用于靶向p53/MDM2相互作用,其中部分分子表现出更好的细胞通透性。研究利用乳酸脱氢酶释放实验评估膜完整性,并通过对照实验验证细胞内p53的激活情况。结果显示,一些订书肽能够激活p53且不引起膜破坏,而另一些则导致细胞泄漏。总体来看,“缝合”肽可能因更高的疏水性和构象刚性而具有更好的细胞通透性,但在某些情况下,即使疏水性和结构稳定性增加,也可能导致通透性下降,说明疏水性对细胞进入的影响具有更复杂的规律。
总体而言,引入构象约束通常能够改善肽的细胞摄取行为,但序列与细胞通透性之间的关系十分复杂。在设计具有良好细胞通透性的构象受限肽时,可以将总体疏水性、适度的正电荷、较高的α螺旋含量以及连续的疏水与带电表面作为重要参考因素。同时,细胞类型也会显著影响摄取效率,例如在巨胞饮作用上调的癌细胞中往往观察到更高的摄取水平。需要注意的是,许多研究使用荧光标记肽进行分析,而荧光标签可能改变分子行为。未来,利用更高通量的检测方法(例如氯代烷渗透实验)、能够快速生成多样化受限肽库的合成技术以及机器学习方法,有望进一步推动这一领域的发展。与此同时,对细胞穿透肽和天然环肽的大量系统研究也为未来设计靶向PPI的构象受限肽提供了重要启示。
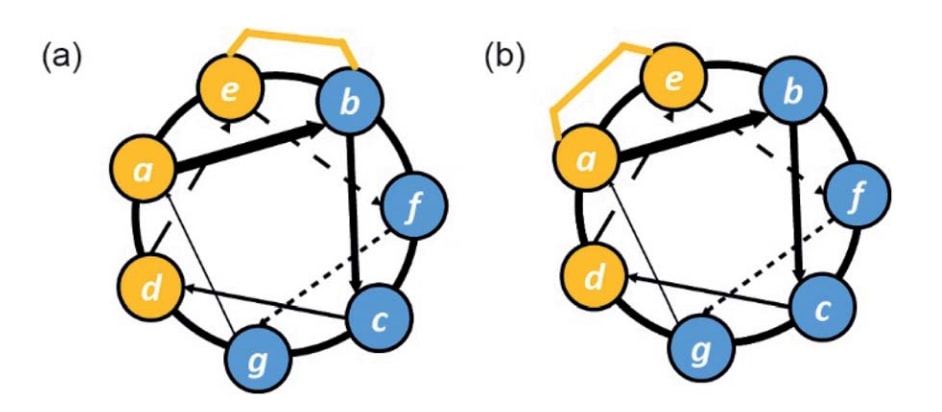
图10|螺旋轮图示展示订书位置的细微差异。 (a)订书位于疏水表面与亲水表面的边界位置,这通常通过扩大疏水表面积而提高细胞摄取能力;(b)订书形成连续疏水表面,从而可能进一步提高细胞摄取能力。图中橙色表示疏水残基及订书结构,蓝色表示亲水或带正电荷的残基。
4 总结
在过去二十年中,各类合成构象约束方法的发展显著推动了用于干扰传统上被认为“难以成药”的蛋白质-蛋白质相互作用(PPIs)的肽类分子的发现。与天然肽相比,构象受限肽通常被认为具有更高的结合亲和力、相似的结合模式以及更好的细胞摄取能力。此外,虽然该研究未进行详细讨论,但构象受限肽还常表现出更高的抗蛋白酶稳定性,这一优势已在大量研究中得到充分证实。
近年来的研究结果表明,这一领域仍存在许多隐藏的复杂性。该研究总结的研究进展显示,构象受限肽的理性设计框架正在不断发展。在设计过程中,需要综合考虑结合机制与热力学特征、约束结构与蛋白之间可能存在的直接相互作用、约束对结合态与游离态构象的细微结构影响,以及决定细胞摄取能力的复杂序列-结构空间。除此之外,构象受限肽在体内分布、代谢、清除、免疫原性以及其他药代动力学/药效学(PK/PD)性质等方面的系统研究仍然较为有限。
尽管如此,构象受限肽已经在很大程度上克服了线性肽配体在应用中的诸多限制,并推动了一些候选分子进入临床试验。随着构象受限肽被用于靶向越来越广泛的生物学靶点,并逐渐整合到蛋白降解靶向嵌合体(PROTAC等)等新型策略中,这类分子预计将在化学生物学与药物发现研究中持续发挥重要作用。