Chemical Reviews 2025 | 难以成药靶标的计算机辅助药物发现

Sun, Q.; Wang, H.; Xie, J.; Wang, L.; Mu, J.; Li, J.; Ren, Y.; Lai, L. Computer-Aided Drug Discovery for Undruggable Targets. Chem. Rev. 2025, 125 (13), 6309–6365. https://doi.org/10.1021/acs.chemrev.4c00969.
0 摘要
难以成药靶标是指具有重要治疗意义但难以通过传统药物设计方法进行有效干预的一类靶标。这类靶标通常具有一些特殊特征,例如结构高度动态、缺乏明确的配体结合口袋、活性位点高度保守,以及其功能主要通过蛋白质−蛋白质相互作用进行调控。近年来,计算模拟技术和人工智能的发展正在深刻改变药物设计的研究格局,并催生出多种新的策略以克服这些困难。
该综述综述了针对难以成药靶标开展药物设计的最新计算方法进展,并结合若干成功案例进行说明,同时讨论当前仍然存在的挑战以及未来的发展方向。 重点关注四类主要靶标类型:内在无序蛋白、蛋白质别构调控、蛋白质−蛋白质相互作用以及蛋白降解机制,同时也对新兴靶标类型进行了介绍。
此外,还讨论了人工智能驱动方法如何改变这一领域的发展,从蛋白质−配体复合物结构预测和虚拟筛选,到针对难以成药靶标的从头配体生成等方面均取得显著进展。计算方法与实验技术的进一步整合有望带来新的突破,从而克服难以成药靶标带来的挑战。随着该领域持续发展,这些进展有望拓展可成药靶标空间,为过去难以治疗的疾病提供新的治疗机会。
1 引言
表型变化的观察在历史上一直是发现新型治疗药物的重要途径。尽管表型筛选在药物发现中仍然发挥着关键作用,但在理解疾病机制与开发有效治疗手段之间仍然存在显著的知识鸿沟。随着分子生物学的发展,以靶标为中心的策略逐渐兴起,并深刻改变了药物发现的研究模式。在这一策略中,靶标识别和靶标验证是两个至关重要的步骤。 “药物靶标”通常指在疾病发生和发展过程中发挥关键作用,并能够通过分子调控实现治疗干预的生物大分子。
随着对疾病机制研究的不断深入以及人类基因组计划的完成,研究者已经识别出数百个潜在药物靶标。然而,从潜在靶标到经过验证的药物靶标之间仍然存在较长的发展过程。 根据Therapeutic Target Database的统计,目前2895种已获批准的药物仅对应532个成功验证的药物靶标。 大多数已验证的药物靶标属于若干蛋白质家族,例如G蛋白偶联受体、核激素受体、蛋白激酶、锌金属肽酶、丝氨酸蛋白酶以及离子通道等。 这些靶标通常可以归纳为三大类:受体、酶以及离子通道。 可成药靶标通常具有以下特点:与疾病进程密切相关、具有明确的配体结合口袋,并且在配体结合后能够产生功能变化。
尽管传统可成药靶标在当前治疗领域中占据主导地位,但超过85%的疾病相关潜在靶标并不符合传统的可成药性标准。 这类靶标通常被称为“难以成药靶标”。这些靶标为传统药物设计方法带来了显著挑战,其典型特征包括以下几个方面:第一,功能界面通常较大且平坦,缺乏典型可成药靶标所具有的深而明确的配体结合口袋;第二,活性位点高度保守,难以设计具有选择性的配体以实现功能调控;第三,内在无序蛋白或包含内在无序区域的蛋白缺乏稳定的三维结构,难以进行传统配体设计;第四,其功能常通过蛋白质−蛋白质相互作用以及复杂蛋白复合物的形成实现;第五,一些疾病抑制蛋白需要通过激活而非抑制来发挥治疗作用。
尽管面临诸多挑战,针对难以成药靶标的药物研究领域仍在迅速发展。越来越多的研究表明,这些靶标蕴含着尚未被充分开发的巨大潜力。随着药物发现技术的进步以及对疾病生物学理解的不断深化,可用于药物开发的靶标范围正在不断扩大。值得注意的是,一些曾被认为难以成药的靶标已经通过创新策略成功实现药物开发,并产生了临床获批的抑制剂。这些突破表明,将难以成药靶标转化为可操作的药物发现机会是完全可能的。
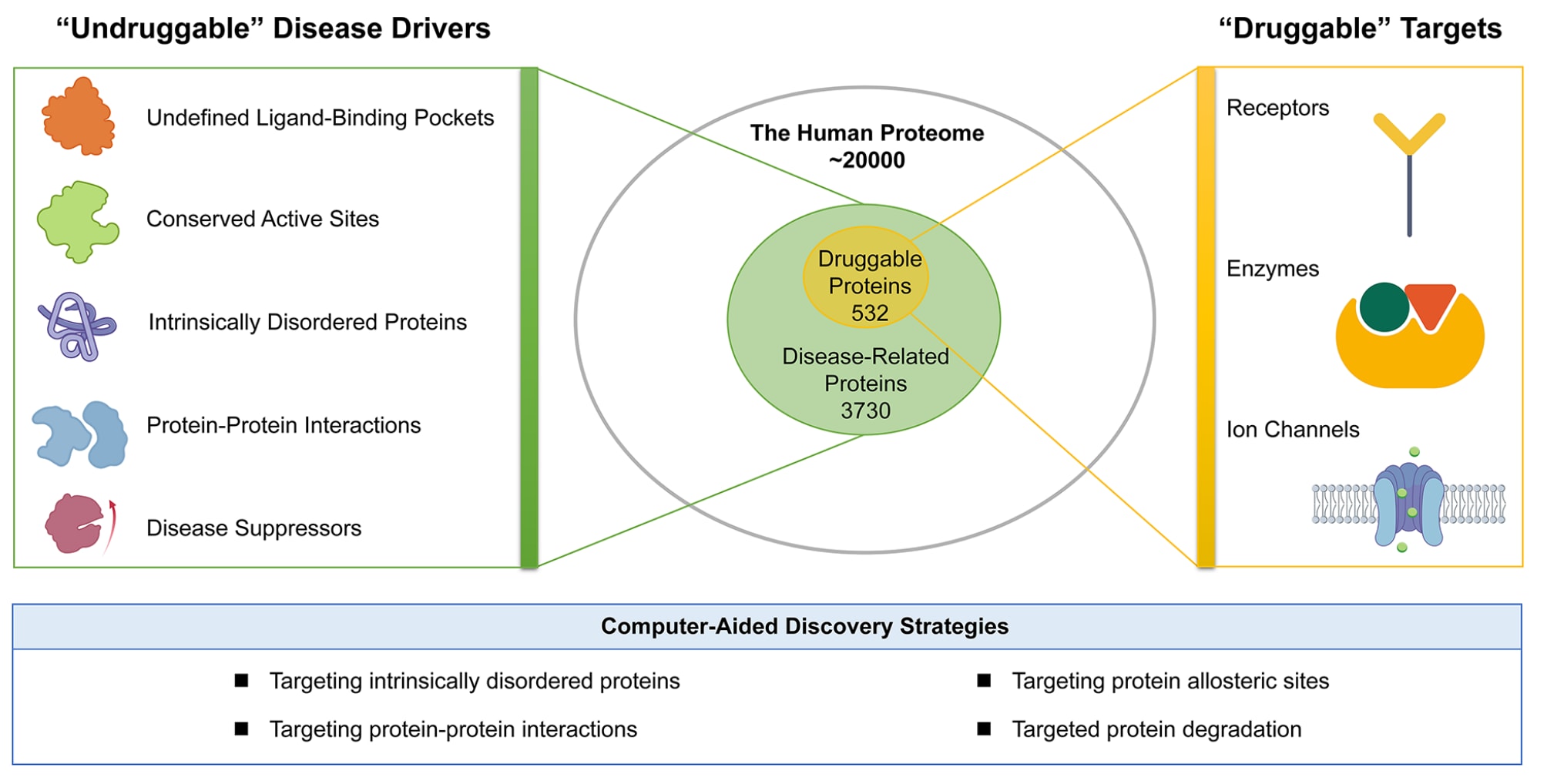
图 1|可成药靶点与不可成药靶点的示意图。 在人类与疾病相关的蛋白质组中,超过 85% 被认为是不可成药(undruggable)的靶点,并且这些靶点通常具有一些共同特征。计算机辅助药物开发策略能够显著提升针对不可成药靶点的药物发现过程。
1.1 “难以成药”靶标的分类
难以成药靶标为药物发现提供了重要机遇,因为许多蛋白质在疾病的发生和发展过程中具有明确的功能。通常可以将难以成药靶标划分为以下五类。随着新型药物发现技术的发展,一些原本被认为难以成药的靶标已经逐渐转变为可成药靶标。
第一类是缺乏明确配体结合口袋的蛋白。 例如小GTP酶通常被认为是难以靶向的蛋白家族。RAS是一类参与细胞信号传导的GTP酶家族,包括KRAS、HRAS和NRAS,在多种癌症中经常发生突变,因此成为肿瘤研究中最重要的靶标之一。RAS能够以皮摩尔级亲和力结合其天然底物GTP,这使得竞争性抑制剂的开发极为困难。此外,RAS通过在GTP和GDP结合状态之间转换实现活化与失活。活化后的RAS能够与多种下游效应蛋白相互作用,而这些蛋白界面通常较为平坦,缺乏深层配体结合口袋。 由于其体积较小、表面光滑且对底物具有极高亲和力,RAS长期被认为是难以成药的靶标。直到针对KRASG12C突变的共价抑制剂被开发出来,这一观点才被改变。 Shokat课题组率先发现了一系列能够特异性结合KRASG12C的共价抑制剂,这些分子在KRAS的switch-II区域附近结合,并通过别构机制稳定其失活构象。在此基础上,研究者进一步开发出更高效的共价抑制剂,例如AMG510和MRTX849系列。其中AMG510(Sotorasib)和MRTX849(Adagrasib)已获临床批准,用于治疗携带KRASG12C突变的晚期或转移性非小细胞肺癌和结直肠癌。目前,研究者已经开发出多种RAS抑制剂,包括KRASG12C特异性共价抑制剂、针对KRASG12D的疗法以及泛RAS抑制剂等。
第二类是活性位点高度保守的蛋白。 磷酸酶是一类催化蛋白去磷酸化的酶,与激酶在功能上互为对应。根据作用方式不同,磷酸酶可以分为蛋白丝氨酸/苏氨酸磷酸酶和蛋白酪氨酸磷酸酶。越来越多的证据表明,多种磷酸酶与癌症等疾病密切相关,因此具有潜在治疗价值。然而,磷酸酶长期被认为难以成药,其原因主要包括两个方面:一是其催化位点高度保守,难以在同源蛋白之间实现选择性抑制,从而可能产生毒性;二是催化位点通常带正电荷,更倾向结合带负电的分子,导致候选分子的细胞通透性和生物利用度较差。 含Src同源结构域2的蛋白酪氨酸磷酸酶2(SHP2)是PTP家族研究最为深入的成员之一,并被认为是潜在的抗癌靶标。 近年来,针对SHP2的别构抑制剂的开发为这一长期被忽视的酶家族带来了新的希望。目前已有多个针对PTP和PSTP家族的抑制剂进入临床试验阶段,但尚未有药物获得临床批准。
第三类是内在无序蛋白。 内在无序蛋白或包含内在无序区域的蛋白在生理条件下缺乏稳定的三级结构。尽管结构高度灵活,这类蛋白在多种疾病中发挥重要作用,包括癌症、神经退行性疾病和糖尿病,并且约占人类蛋白质组的40%。与折叠蛋白相比,内在无序蛋白通常具有较低的序列复杂度、较少的大型疏水残基、较多的带电和极性残基以及更高的结构柔性。这些特征使其难以通过传统药物设计方法进行靶向,因此被视为难以成药靶标。转录因子是一类能够结合特定DNA序列并调控基因表达的蛋白,在多种细胞功能中发挥关键作用。在约300个与疾病表型相关的转录因子中,只有少数成功被小分子药物靶向。其中一个重要原因是大多数转录因子主要由内在无序结构组成。转录因子c-Myc是著名的致癌蛋白,参与多条关键的信号通路,其异常表达在多种癌症中普遍存在,因此被认为是重要的药物靶标。然而,由于其属于内在无序蛋白,目前尚无针对c-Myc的获批药物。随着对内在无序蛋白结构与功能认识的加深以及计算方法的发展,针对c-Myc的新策略正在出现。例如小型蛋白Omomyc以及肽类分子IDP121已经进入临床研究阶段。
第四类是蛋白质−蛋白质相互作用靶标。 蛋白质−蛋白质相互作用是维持细胞生命活动的重要基础,因此调控这类相互作用在癌症、感染性疾病以及神经退行性疾病治疗中具有重要潜力。然而,由于蛋白质−蛋白质相互作用界面通常较大、较平坦且极性较强,小分子药物很难有效结合这些界面。 因此,PPI调节剂往往比典型口服药物分子更大,这可能导致较差的药物性质。 随着计算化学和结构生物学的发展,针对PPI界面的理性设计取得了显著进展。目前已经开发出多种PPI调节剂,其中一些已进入临床试验阶段。一个典型例子是抗凋亡蛋白Bcl-2家族,其能够与促凋亡蛋白Bax和Bak结合。阻断Bcl-2与Bak或Bax之间的相互作用可以防止肿瘤细胞逃避凋亡。另一类重要靶标是XIAP蛋白,它是caspase-3、7和9的强效抑制剂,并在多种癌症中高表达。线粒体来源的caspase激活因子Smac能够与XIAP的BIR结构域结合,从而阻止XIAP与促凋亡蛋白相互作用并促进细胞凋亡。模拟Smac结构的XIAP拮抗剂目前已作为潜在抗癌药物进入临床试验。此外,RAS和p53等著名靶标同样涉及蛋白质−蛋白质相互作用。
第五类是疾病抑制蛋白。 这类靶标在疾病中通常表现为功能缺失,因此需要药物激活其功能,而不是抑制其活性,这使药物设计更加困难。TP53是人类癌症中突变频率最高的基因,其编码的p53蛋白是一种肿瘤抑制型转录因子,负责DNA修复和细胞应激反应。当p53因突变或缺失而失活时,细胞关键调控过程受到破坏,从而促进肿瘤发生。目前恢复p53功能的策略主要包括阻断野生型p53与其负调控因子的相互作用,以及重新激活突变型p53。基于这些策略,已有多种小分子进入临床研究阶段。
1.2 从“难以成药”到“可成药”
随着药物发现策略的不断进步,“难以成药”这一概念正逐渐被更为乐观的表述所取代,例如“难以靶向”或“尚未被靶向”。 值得注意的是,一些曾被认为难以成药的靶标,如Bcl-2和RAS,已经成功开发出临床获批的抑制剂,因此不再被视为难以成药靶标。 针对复杂靶标的创新药物设计策略正在不断出现。
近年来,计算工具和人工智能技术的突破显著提高了这些策略的可行性,并推动了多个关键研究方向的发展,例如药物靶标识别、蛋白结构预测、虚拟筛选加速、从头药物设计以及疾病发生机制研究。基于人工智能的蛋白结构预测方法,例如AlphaFold和RoseTTAFold,在预测生物分子复合物结构方面已经取得了极高的准确度。
该综述主要总结针对难以成药靶标开展药物设计的计算方法,并讨论未来面临的挑战。重点关注近年来计算策略的发展,并系统介绍这一领域中采用的主要方法。同时,通过若干最新研究案例展示这些方法的可行性与潜力。为了形成清晰的研究框架,相关进展将根据靶标类型进行分类,包括针对内在无序蛋白、别构位点、蛋白质−蛋白质相互作用以及蛋白降解机制的药物设计策略。
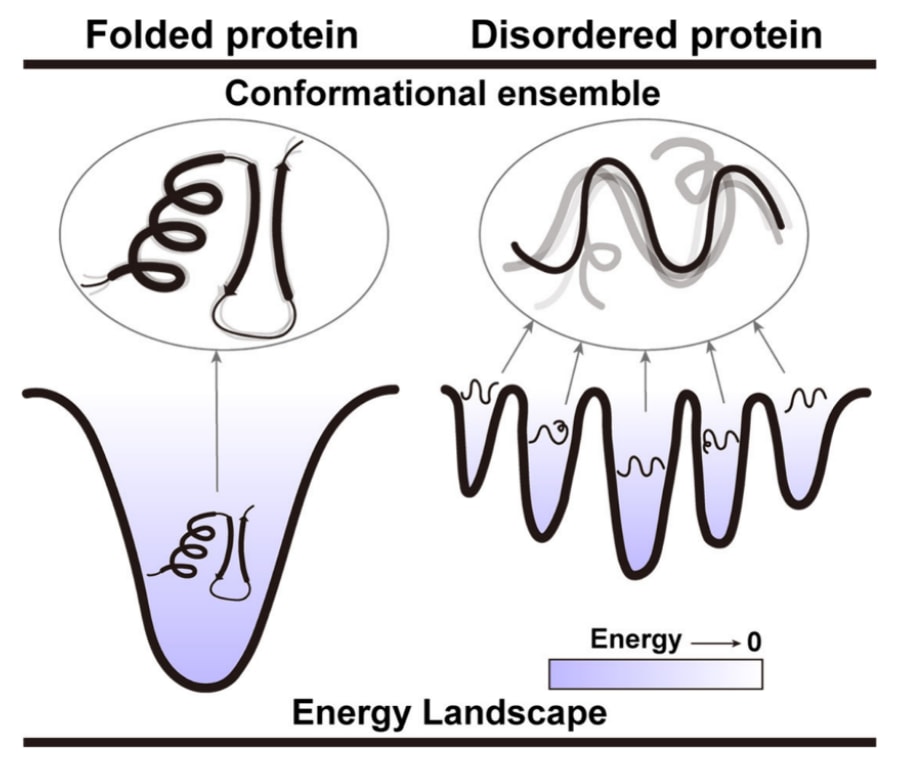
图2|折叠蛋白与内在无序蛋白的示意图。 完全折叠蛋白具有单一且较深的漏斗状自由能景观,因此其构象基本稳定在最低能量状态。相比之下,内在无序蛋白(IDP)在多个低能量构象之间仅存在较小的能垒,使其能够在不同构象之间持续转换,从而形成一种动态且异质的构象集合。
2 靶向内在无序蛋白的药物设计
2.1 IDPs作为药物靶点
2.1.1 IDPs作为药物靶点的生物学基础与临床意义
内在无序蛋白(intrinsically disordered proteins,IDPs)及其内在无序区域(intrinsically disordered regions,IDRs)在生理条件下通常缺乏稳定且明确的三维结构。如图2所示,与完全折叠蛋白具有深漏斗形的自由能景观不同,IDPs的自由能景观由多个浅漏斗组成,这些漏斗之间仅由较小的能垒分隔。因此,IDPs并非处于单一稳定构象,而是以多种异质构象构成的动态集合体形式存在,并能够持续、自发地相互转化。这种高度动态的构象集合特性主要由其氨基酸序列所决定。
在序列组成上,IDPs通常富含极性残基,例如R、Q、S、E和K,而促进有序结构形成的残基(I、L、V、W、Y、F和C)则相对较少。 大量极性残基会增强分子内的静电排斥作用并提高与水分子的相互作用,而疏水残基的缺乏则阻碍稳定疏水核心的形成,两者共同 导致蛋白难以维持稳定折叠结构。
IDPs在生物体中广泛存在,并在细胞功能中发挥重要作用。 它们参与多种关键细胞过程,包括转录调控和信号传导等。例如,多数转录因子由于含有无序的转录激活结构域(transactivation domain,TAD),因此被归类为IDPs。IDPs常被视为相互作用网络中的“枢纽”分子。其动态结构使其在与天然蛋白配体结合时能够形成互补构象,从而实现高特异性但相对较低亲和力的结合模式。这种特性使其能够在细胞内通过精确的时空调控与多种不同蛋白发生相互作用,从而在蛋白质相互作用网络中处于中心位置。
此外,IDPs还能通过多价相互作用形成无膜细胞器(membraneless organelles),从而富集或隔离特定分子,进而促进或抑制相关生化反应。
IDPs功能失调与多种重大人类疾病密切相关,因此成为重要的潜在药物靶点。在癌症、心血管疾病以及糖尿病中均可观察到IDPs的广泛存在。IDPs同样参与多种神经退行性疾病的发生。例如,完全无序的α-突触核蛋白(α-synuclein,α-syn)是一种神经元蛋白,参与突触小泡调控及神经递质释放。当α-syn发生异常聚集时会形成具有毒性的淀粉样纤维,这是帕金森病的重要病理特征。因此,抑制α-syn的异常聚集被认为是一种具有潜力的治疗策略。
在癌症治疗领域,转录因子尤其是致癌蛋白和肿瘤抑制因子,由于在多数人类癌症中存在过度激活或异常抑制现象,被视为极具价值的治疗靶点。例如转录因子c-Myc是一种全长无序的致癌蛋白。泛癌分析显示,在来自33种癌症类型的约9000个样本中,MYC基因在21%的样本中发生扩增。在依赖MYC致癌基因的癌症中,癌细胞的存活与增殖依赖于MYC持续的过表达。c-Myc通过多种机制促进肿瘤发生,包括促进细胞增殖、抑制细胞衰老以及帮助肿瘤逃避免疫监视等,因此成为研究最为广泛的药物靶点之一。
另一重要例子是转录因子p53。 p53是一种含有无序转录激活结构域的肿瘤抑制蛋白,在超过50%的癌症中发生突变或失活。p53失活的主要原因之一是其负调控因子MDM2的异常抑制作用。p53与MDM2的结合位点位于其无序TAD区域。由于该区域缺乏稳定结构,相关药物设计通常通过开发MDM2抑制剂以间接激活p53。然而目前的临床研究表明,MDM2抑制剂的治疗效果有限,且容易诱导p53产生新的突变。
雄激素受体(androgen receptor,AR)同样是一个具有无序TAD结构域和折叠配体结合结构域(ligand-binding domain,LBD)的转录因子,在前列腺癌细胞的生长和存活中发挥关键作用。 LBD中含有能够结合雄激素的配体结合口袋,雄激素结合后会诱导AR转位至细胞核并作为转录激活因子发挥功能。当前临床上广泛采用与AR-LBD结合的配体(例如恩杂鲁胺Enzalutamide)来抑制AR功能,从而阻断雄激素刺激以治疗前列腺癌。然而,LBD缺失等突变会导致药物耐受,使AR在无雄激素条件下仍能被激活,进而形成去势抵抗性前列腺癌(castration-resistant prostate cancer,CRPC)。因此,近年来大量研究开始尝试开发能够直接结合AR-TAD并抑制其功能的药物。
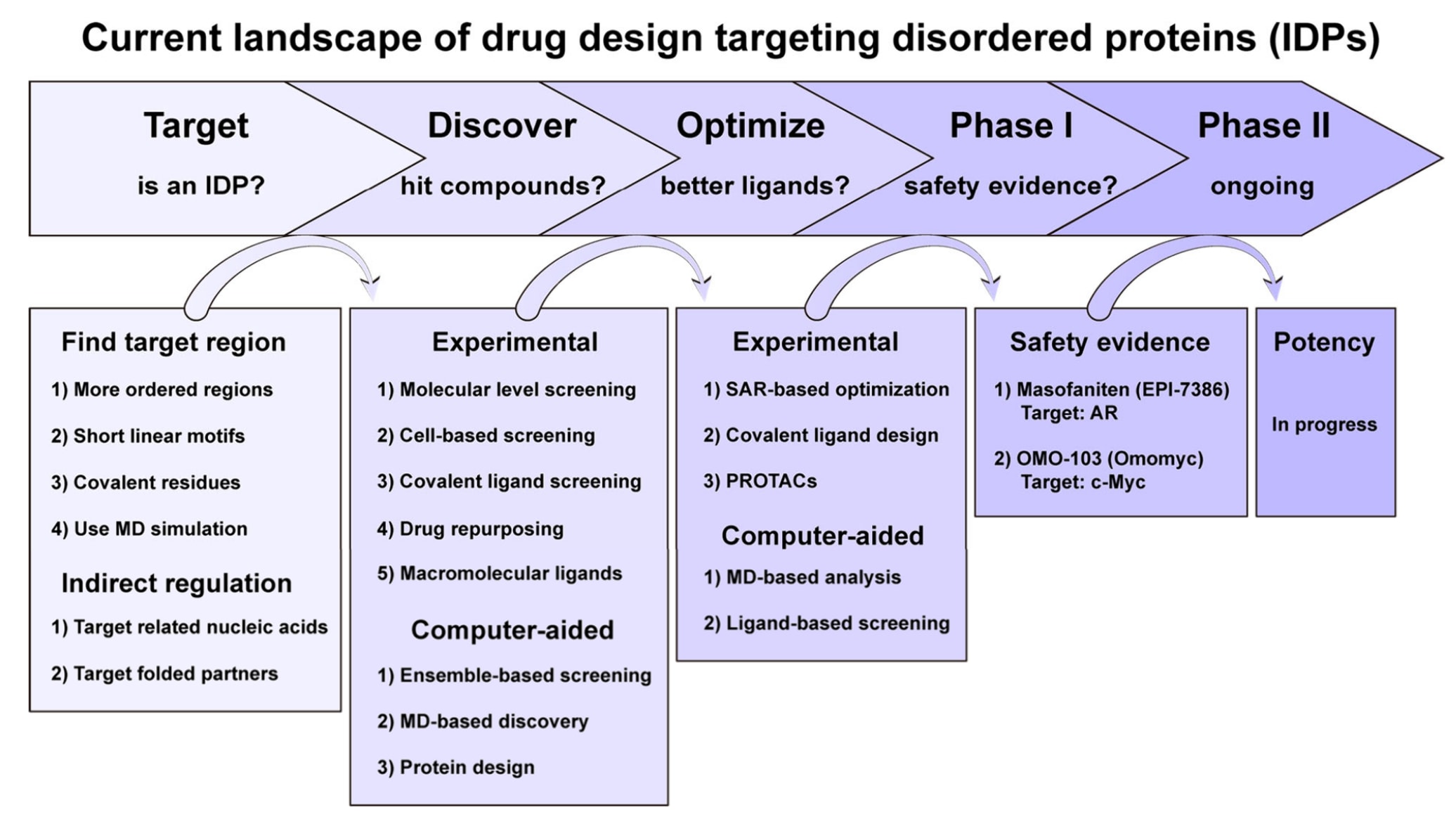
图3|靶向内在无序蛋白的药物设计研究现状。 EPI-7386和Omomyc的Ⅰ期临床试验结果已在相关文献中进行了报道。缩写:MD,分子动力学;SAR,结构–活性关系;PROTACs,蛋白降解靶向嵌合体。
2.1.3 IDPs作为药物靶点的研究现状
尽管过去十余年中针对c-Myc、AR等靶点开展了大量研究,目前仍没有直接靶向内在无序蛋白(IDPs)的药物获批上市,这凸显了IDP药物设计所面临的巨大挑战。 然而,随着计算方法与实验技术的发展,研究者对IDP序列—构象集合—功能之间关系的理解不断加深,同时药物设计策略也持续进步,IDP相关药物的开发正逐渐成为现实(图3)。
为了克服IDPs缺乏稳定结构所带来的困难,研究中常采用间接调控策略,例如靶向其在DNA上的转录调控位点、mRNA或其具有稳定结构的相互作用伙伴等。对于直接靶向IDP的研究,多种药物发现策略被用于筛选具有初始活性的候选化合物。其中,基于计算机辅助的构象集合药物发现方法(ensemble-based drug discovery,EBDD)在效率和成本方面具有明显优势。
目前,通过分子动力学(MD)模拟结合实验研究,已经初步获得了IDP与配体结合特征的相关认识。然而,活性更高的配体仍主要依赖于结构—活性关系(structure–activity relationship,SAR)分析进行优化。值得关注的是,针对c-Myc的配体Omomyc(OMO-103)以及针对AR的配体EPI-7386(Masofaniten)在近期Ⅰ/Ⅱ期临床试验中显示出良好的安全性和疗效,表明直接靶向IDPs在临床上具有可行性,也进一步推动了IDP药物开发领域的发展。
尽管如此,在IDP药物设计过程中,高效的配体优化仍然是一项重要挑战。未来仍需要发展更先进的计算方法以解析IDP—配体的动态结合模式,同时还需要构建具有更高空间与时间分辨率的实验技术,从而更深入地理解IDP相关的分子机制并促进药物设计。
2.2 IDPs的构象集合
在开展基于结构的IDP药物设计时,获取其构象集合是关键步骤之一。尽管核磁共振(NMR)和小角X射线散射(SAXS)等实验方法能够提供IDP的动态结构特征,目前用于采样IDP构象集合的主要手段仍然是分子动力学(MD)模拟。近年来针对IDP模拟的专用力场取得了显著进展,然而IDP高度柔性的特点以及缺乏稳定结构的性质,使传统MD模拟在高效探索其广阔构象空间时面临挑战。为了解决这一问题,研究中逐渐采用增强采样方法和粗粒化(coarse-grained,CG)力场。同时,随着深度学习技术的发展,基于人工智能的方法也逐渐成为直接生成IDP构象集合的一种潜在替代策略。
2.2.1 IDP专用力场
由于IDP具有极高的柔性,其构象状态分布更加广泛,并且更容易陷入局部能量陷阱,传统针对折叠蛋白开发的经典力场往往难以准确描述IDP的构象特性。因此,研究者开发了一系列针对IDP的专用力场。这些力场的改进主要集中在三个方面:调整二面角参数、引入CMAP修正以及优化蛋白质与水之间的相互作用。
一种常见策略是重新调整二面角参数,尤其是主链二面角ff03*和ff99SB*分别基于ff03和ff99SB发展而来,其参数优化借助Lifson-Roig螺旋-线圈理论完成。相较于ff03,ff03*往往高估螺旋含量,而ff99SB*则相对于ff99SB低估螺旋含量,二者均配合TIP3P水模型使用。随后提出的ff03w在ff03*基础上结合TIP4P/2005水模型进一步改进。类似地,OPLS-AA/M和OPLS3通过重新参数化主链和侧链二面角(χ₁和χ₂),利用从头计算获得的扭转能扫描数据进行优化。其中OPLS-AA/M在模拟含脯氨酸和甘氨酸的肽段时表现良好,而OPLS3在蛋白-配体结合模拟中具有较高准确性。CHARMM22*是CHARMM22的重新拟合版本,重点优化蛋白折叠与解折叠过程,并在模拟villin headpiece蛋白时表现出良好性能。此外,还出现了采用残基特异性二面角参数的力场,例如RSFF1和RSFF2,分别基于OPLS/AA和ff99SB发展而来。RSFF2在RSFF1基础上进一步改进,解决了α螺旋和β折叠稳定性被高估的问题。
另一类策略通过引入CMAP参数对力场进行修正。例如,CHARMM22/CMAP(即CHARMM27)在CHARMM22基础上平衡了螺旋和无规线圈构象,但在稳定发夹结构方面表现不佳,并且在α-syn模拟中存在螺旋含量过高的问题。随后提出的CHARMM36通过引入NMR数据进行优化,提高了螺旋与发夹结构形成的协同性,同时保持了对折叠蛋白的良好描述能力。然而在某些IDP模拟中,CHARMM36会产生过多左手螺旋结构,这一问题在后续提出的CHARMM36m中通过残基特异性的CMAP策略得到改进。类似地,a99SB-disp通过优化扭转项和非键相互作用参数,在IDP与折叠蛋白模拟之间取得较好的平衡,但在模拟Aβ肽聚集时仍存在一定局限。
早期的残基特异性CMAP力场,例如ff99IDPs和ff14IDPs,主要针对促进无序结构的氨基酸(G、A、S、P、R、Q、E和K)修改CMAP势能。后续发展出的ff14IDPSFF和CHARMM36IDPSFF则将CMAP修正扩展至全部20种氨基酸。这些力场在长时间尺度模拟中表现良好,并能够较好再现实验NMR观测结果。OPLSIDPSFF也是一种残基特异性力场,通过修正20种氨基酸的主链扭转项并结合TIP4P-D水模型获得良好性能。
ff03CMAP在训练集中同时包含IDP和折叠蛋白结构,强调训练模型选择的重要性,并在TIP4P-Ew和TIP4P-D水模型下表现良好。RSFF2C进一步将CMAP修正扩展至侧链二面角
由于IDP缺乏稳定的疏水核心,蛋白质与水之间的相互作用在模拟中尤为关键。早期IDP力场往往低估回转半径(radius of gyration,
部分力场还直接修改蛋白-水相互作用项。例如ff03ws在ff03w和TIP4P/2005基础上发展而来,通过减弱蛋白-蛋白相互作用来解决其过度稳定化问题,这一问题在IDP聚集中尤为突出。类似地,a99SB-UCB通过修改非键参数和主链参数对这一问题进行了改进。CHARMM36m则同时调整了水分子中氧和氢原子的参数,以提高IDP模拟的准确性。
总体而言,IDP专用力场的发展显著提高了MD模拟的准确性,为获得更加可靠的构象集合提供了基础,并为后续虚拟筛选以及IDP-配体相互作用机制的研究提供了重要支持。
表1|代表性增强采样方法在内在无序蛋白(IDP)研究中的典型应用实例。

2.2.2 IDP构象的增强采样
增强采样方法旨在加速体系跨越能量势垒,从而提高构象采样效率。实现这一目标通常可以通过多种策略,例如引入偏置势、修改势能景观、改变有效温度、人工筛选能够逃离能量陷阱的构象,或将多种策略进行组合。
在引入偏置势的方法中,偏置能量的定义至关重要。其中最常见的策略是利用集体变量(collective variables,CVs)作为偏置势。例如在元动力学(metadynamics,MTD)中,会加入依赖于历史轨迹的偏置势,从而避免体系重复访问已采样的状态,并逐渐使自由能景观趋于平坦。随着基于预设CVs的偏置能量不断累积,IDPs能够更容易跨越能量势垒。类似地,在伞形采样(umbrella sampling,US)中,集体变量被划分为多个相互重叠的“窗口”,并在每个窗口中独立进行采样。
基于CV的方法还可以结合并行模拟技术进一步增强,例如复制交换伞形采样(replica-exchange umbrella sampling,REUS)、偏置交换元动力学(bias-exchange metadynamics,BEMD)、并行温度元动力学(parallel tempering metadynamics,PTMTD)以及并行偏置元动力学(parallel bias metadynamics,PBMetD)。这些方法已被广泛应用于IDP构象集合的采样。例如MTD方法被用于研究WH5肽的螺旋-线圈转变以及p53与MDM2之间耦合折叠与结合过程;偏置交换MTD则被用于研究pKID与KID蛋白之间的耦合结合机制。此外,MTD方法还被用于探索IDP构象转变路径。表1总结了增强采样方法在IDP模拟中的典型应用实例。
然而,基于CV的方法存在一个重要局限,即必须事先定义合适的CV,这通常需要对研究的IDP体系具有较深入的了解,并且CV的选择质量会显著影响采样效率。为了解决这一问题,研究者开始利用人工智能模型辅助选择CV。早期研究主要使用线性降维方法,例如主成分分析(principal component analysis,PCA)和多维尺度分析(multidimensional scaling,MDS)。随后的一些研究采用深度学习模型,在非线性潜在空间中表示能量景观。
无CV方法则无需预先定义集体变量,因此在模拟IDP这种高维构象波动体系时具有一定优势。这类方法通常通过基于温度调节(tempering)的策略使能量景观变得平坦,其核心思想是提高体系的有效模拟温度。典型方法包括temperature-cool walking(TCW)、annealed importance sampling以及simulated tempering等。近年来,这类基于温度调节的方法常与复制交换技术结合使用,例如温度复制交换分子动力学(temperature replica-exchange molecular dynamics,TREMD)。此外还发展出一些变体方法,例如溶质温度复制交换(replica exchange of solute temperature,REST)和溶剂温度复制交换(REST2)。
REST和REST2可被视为REMD的简化版本,其自由度较少。该类方法的核心思想是将体系划分为“溶质”和“溶剂”两个区域,并仅在溶质或溶剂区域中修改温度或Lennard-Jones势能参数。这类方法已被广泛用于IDP构象集合采样。例如Ngo等利用REMD模拟了Aβ42四聚体结构,Das等利用REMD研究了人胰岛淀粉样多肽(hIAPP)的聚集过程,Hu等则结合AlphaFold2与REMD模拟阐明了蛋白4.1G与NuMA中IDP区域之间的识别机制。在REST方法方面,Kamiya等利用广义REST(gREST)研究了β发夹结构和Trp-cage的折叠转变,Brown等利用REST分析了n16N无序肽的构象集合。REST2是一种较新的方法,目前也已有多种应用。例如Appadurai等同时使用REST和REST2研究His-5蛋白,Reid等则利用REST2模拟PLP、TP2和ONEG肽的结构行为。值得注意的是,REST与REST2之间仍存在一定争议,部分研究认为REST在某些情况下可能优于REST2。然而,复制交换类方法通常需要较高的计算资源,往往需要数百个CPU或多个GPU才能实现高效计算。
另一类增强采样方法通过加入提升势(boost potential)以降低能量势垒,例如加速分子动力学(accelerated molecular dynamics,aMD)。为了解决重加权过程中产生的能量噪声问题,随后提出了高斯加速分子动力学(Gaussian accelerated MD,GaMD)。GaMD通过引入新的谐和提升势,并结合新的重加权方法,能够较为准确地恢复自由能景观。其衍生方法包括ligand-GaMD和peptide-GaMD,分别被用于研究分子配体结合以及高度无序肽的动态行为。GaMD也被应用于IDP体系研究。例如Alcantara等利用GaMD研究富含脯氨酸的无序序列中的异构化过程,Gunasinghe等则利用GaMD采样c-Myc蛋白的构象集合。
随着机器学习技术的发展,研究者开始尝试将其整合到采样流程中。这类方法通常遵循一个循环流程:首先进行短时间MD模拟;随后利用机器学习方法描述轨迹分布;然后在构象分布空间中识别“边缘”构象;将这些构象作为新的初始结构开展新的短时间模拟;最后不断重复这一过程。例如Shkurti等提出的互补坐标分子动力学(complementary coordinates MD,CoCo-MD)利用COCO方法从IDP模拟轨迹中筛选代表性结构,该方法最初用于分析NMR构象集合。另一个例子是Zhang等提出的frontier expansion sampling(FEXS)。类似的工作还包括structural dissimilarity sampling(SDS)、self-avoiding conformational sampling(SACS)、parallel cascade selection molecular dynamics(PaCS-MD)、nontargeted PaCS-MD、anomaly detection PaCS-MD以及independent nontargeted PaCS-MD等。
这类方法面临的主要挑战在于,随着迭代次数增加,降维计算所需时间可能超过MD模拟本身的时间成本,从而影响实际应用效率。因此,尽管这些方法未必会成为IDP增强采样的主流策略,但在某些特定研究中仍然取得了成功应用,相关实例见表1。
当模拟中引入偏置势时,通常需要进行重加权处理以恢复无偏样本并重建准确的自由能面。常用方法包括针对具有特定CV偏置模拟的加权直方图分析方法(weighted histogram analysis method,WHAM),以及更为通用的多状态Bennett接受比(multistate Bennett acceptance ratio,MBAR)。对于无偏MD模拟轨迹,还可以采用基于构象群体(population-based)的重加权方法进行分析。
2.2.3 降低IDP体系的计算复杂度
在某些研究情境中,并不一定需要获得IDP构象集合的完整全原子细节信息,研究重点往往更侧重于IDP体系的整体性质。此外,在蛋白聚集或生物分子凝聚体体系中,体系所包含的原子数量往往非常庞大。尽管已有研究利用全原子力场对包含多条IDP链的完整凝聚体体系进行模拟,但这种模型通常需要进行一定程度的简化,以降低原子层级复杂度。目前常见的两类方法包括隐式溶剂模型和粗粒化(coarse-grained,CG)力场,其核心思想是减少体系自由度,从而降低计算复杂度。凭借这一优势,两类方法均已广泛应用于大规模IDP体系研究。
隐式溶剂模型通过直接估算溶剂化自由能来描述溶剂对溶质热力学性质的平均影响。除了降低计算复杂度之外,隐式溶剂模型还具有另一个重要优势:由于消除了溶剂摩擦带来的黏滞效应,体系采样速度得以提高,从而提升构象空间探索效率。此外,去除溶剂自由度还能减少REMD模拟所需的复制数,显著降低计算成本。常见的蛋白模拟软件包,如CHARMM和Amber,已经内置多种隐式溶剂模型,例如Amber中的GB模型(GB-HCT、GB-OBC和GBNeck)以及CHARMM中的GB模型(GBSW、GBMV和GBMV2)。随后的一些研究进一步针对IDP体系优化这些模型。例如GB-Neck2模型通过优化多种蛋白体系的溶剂化能,提升了无序肽模拟性能。另一典型模型是ABSINTH模型,其目标是更好地再现肽链的聚合物特性,并建立在EEF1溶剂模型基础之上。ABSINTH已被成功应用于多种IDP研究,例如Aβ肽聚集过程以及IDR序列与构象之间关系的研究。随后提出的ABSINTH-C模型通过引入残基特异性的修正项解决了原ABSINTH模型中Ramachandran分布过于平坦的问题。
相比之下,更加常见的降低计算复杂度方法是采用粗粒化力场。在这类模型中,多个原子被简化为一个刚性珠粒进行表示。对于大型IDP体系而言,CG力场不仅减少了需要模拟的自由度数量,还允许使用比全原子模拟更大的时间积分步长。因此,许多CG模型在计算效率上可比全原子模拟提高数个数量级,成为非常有效的替代方案。
粗粒化模型可以在不同分辨率层级上构建。例如MARTINI模型将每个氨基酸残基表示为多个珠粒,在采样效率与精度之间取得了较好平衡。针对IDP体系,MARTINI-IDP模型通过调整非键Lennard-Jones势参数,并结合全原子模拟与实验数据进行参数化。MOFF力场则利用最大熵算法并结合多种实验数据进行优化,已成功应用于IDP模拟研究。另一种针对IDP开发的CG力场是Thirumalai团队提出的SOP-IDP模型,该模型采用两个珠粒表示一个残基,能够准确再现IDP的小角X射线散射(SAXS)实验曲线。AWSEM力场仅保留Cα、Cβ和O三个原子,其IDP版本AWSEM-IDP通过调整与二级结构相关的势能项并引入新的参数项VRg进行了重新参数化。
除了常规MD模拟外,CG力场还可以与增强采样方法结合。例如Chen等提出的多尺度增强采样(multiscale enhanced sampling,MSES)方法是一种无需定义CV的增强采样策略。该方法的核心思想是将全原子模拟与粗粒化模型进行耦合。为配合该方法,Chen团队还开发了混合分辨率模型HyRes。HyRes对主链原子采用全原子描述,而对侧链原子采用CG描述。MSES方法已经被应用于多个IDP体系研究,例如p53-TAD以及具有螺旋结构的IDP体系。
近年来的研究表明,IDPs在蛋白液-液相分离(liquid–liquid phase separation,LLPS)体系中发挥重要作用,并可能成为潜在药物靶点。因此,在这类体系中模拟多条IDP链形成的凝聚态结构尤为关键。尽管MARTINI模型已被用于LLPS体系模拟,但由于体系规模庞大,其计算成本往往较高。更加高效的方法是基于Cα的粗粒化模型,该方法使用主链Cα原子代表整个残基,并常与slab-geometry策略结合使用。
Dignon等首先将slab-geometry方法应用于蛋白LLPS模拟,并提出了两种力场:KH模型和HPS模型。这两种模型均使用Debye–Hückel近似处理长程静电相互作用,但在短程残基间相互作用处理上有所不同。KH模型采用Kim-Hummer近似,而HPS模型则基于残基疏水性尺度进行参数化。随后研究表明,这些模型不仅可以用于IDP LLPS体系模拟,还可以用于单链IDP模拟。
在HPS模型基础上,Best等通过数据驱动方法重新参数化疏水性尺度,提出FB-HPS模型。Mittal团队则通过重新调整疏水性比例因子提出HPS-Urry力场。Das团队进一步指出早期HPS模型低估了阳离子-π相互作用,从而提出HPS+Cation-π力场。Joseph等对现有多种力场进行了系统基准测试,并提出新的Mippi力场。该模型使用Wang-Frenkel势函数描述短程残基相互作用,并通过全原子伞形采样数据拟合,解决了阳离子-π相互作用被低估的问题。
Lindorff-Larsen团队提出CALVADOS力场,该模型采用改进的Lennard-Jones势(Ashbaugh-Hatch势)并通过单链性质进行参数拟合。随后又提出了CALVADOS2和CALVADOS3等优化版本。COCOMO力场及其改进版本COCOMO2则使用
基于slab-geometry的模拟策略目前已被广泛应用于IDP LLPS研究,例如LAF-1蛋白、FUS蛋白、hnRNP A1蛋白以及hnRNP A2蛋白等体系的模拟研究。值得注意的是,Lindorff-Larsen团队利用CALVADOS2力场对人类蛋白质组中所有预测的IDR区域进行了模拟,并由此构建了IDRome数据库。
在LLPS模拟中还存在更低分辨率的方法。例如Pappu团队开发的基于晶格点的模拟方法LASSI(Lattice Simulation of Sticker and Spacer Interactions),通过简化模型有效描述“sticker–spacer”相互作用框架,为研究LLPS行为提供了一种简洁而高效的计算策略。
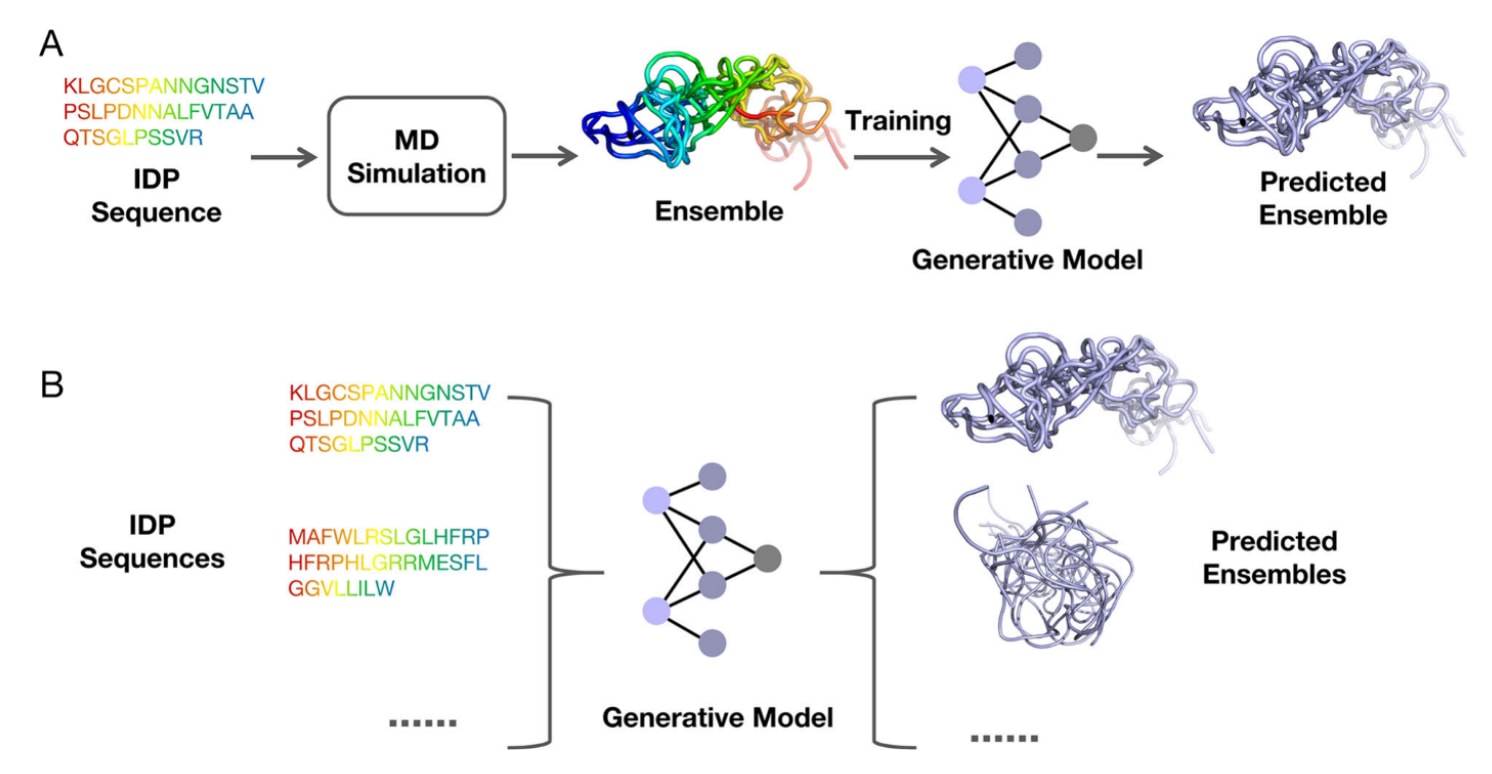
图4|常见的IDP构象生成模型工作流程示意图。 (A)针对特定体系的生成模型;(B)基于序列的生成模型。
2.2.4 基于人工智能的IDP构象集合生成模型
随着深度学习模型的迅速发展,越来越多研究利用深度神经网络挖掘蛋白结构中的潜在特征。这类模型已经取得了一系列重要突破,例如用于蛋白结构预测的AlphaFold2模型以及用于从头蛋白序列设计的proteinMPNN模型。这些成果也成为2024年诺贝尔化学奖的重要科学背景之一。在折叠蛋白结构预测取得成功之后,研究者开始探索类似方法是否能够用于生成内在无序蛋白(IDP)的构象集合。与计算成本较高的分子动力学(MD)模拟相比,深度学习模型在完成训练并配备多个GPU后可以快速生成IDP结构,从而避免长时间MD模拟的计算过程,但同时也对AI模型的精度与效率提出了更高要求。
然而,与折叠蛋白相比,可用于训练的IDP结构数据明显不足。例如截至2024年,蛋白质结构数据库(PDB)中包含超过20万条折叠蛋白结构,而Protein Ensemble Database(PED)中仅包含约500个IDP构象集合。此外,PDB中的结构大多来源于NMR、X射线晶体学以及冷冻电镜(cryo-EM)等实验方法,而PED中的数据则有相当一部分来自MD模拟。因此,无论在数量还是质量上,IDP数据都明显少于折叠蛋白结构数据。尽管存在这些挑战,随着AI生成算法的快速发展,IDP构象集合生成仍然取得了一定进展。目前基于AI的IDP结构生成方法大体可以分为两类:针对特定体系的模型和基于序列的模型。
针对特定体系的生成模型(case-specific models)通常用于生成某一特定IDP的构象集合,每个IDP都需要单独训练模型。这类模型通常以MD模拟轨迹作为训练数据。其基本流程如图4A所示:首先获得目标IDP的初始MD轨迹,随后利用该轨迹训练模型;训练完成后,模型作为生成器用于生成该IDP的构象。这类模型通过潜在特征表示IDP体系的构象空间,并生成比原始训练数据更丰富的构象集合。其思想与AI增强采样方法存在一定相似性,可以视为对这些方法的延伸。不过,该类模型的重要局限在于缺乏可迁移性。一旦IDP序列发生变化,就需要重新进行MD模拟并训练模型,这在大规模应用中成本较高。
Boltzmann Generator是典型的针对特定体系的生成模型之一,该模型采用可逆生成建模技术normalizing flow。虽然目前主要应用于快速折叠蛋白体系,但由于快速折叠蛋白与IDP均具有高度分散的构象空间,理论上也可以扩展到IDP体系。该方法的关键优势在于能够近似Boltzmann分布,其实现方式是通过训练模型将简单先验分布映射到目标Boltzmann分布。不过由于normalizing flow模型表达能力有限,其应用范围仍受到一定限制。
为克服这一问题,研究者提出了其他生成策略。例如Zhou等开发了一种基于自编码器(autoencoder)的模型用于扩展IDP构象空间。研究结果表明,对于Q15、Aβ40和ChiZ等多个IDP体系,该方法生成的构象多样性明显高于原始MD模拟轨迹训练集。随后提出的Phanto-IDP模型则利用变分自编码器(variational autoencoder,VAE)作为生成模型,同样基于MD模拟轨迹进行训练。另一种方法DynamICE则采用不同策略,其目标不是从已有构象集合重新采样,而是直接生成与NMR实验数据一致的结构集合,从而为基于MD数据训练的方法提供补充。
另一类方法是基于序列的生成模型(sequence-based models),其目标是直接根据输入的IDP序列生成构象集合。与针对特定体系的模型相比,这种方法更加接近自动化和通用化的结构预测思想。其基本流程如图4B所示:用户输入IDP序列后,模型直接生成对应的构象集合,类似于AlphaFold或ESMFold等端到端结构预测模型。然而,基于序列的方法也面临显著挑战。首先是高质量训练数据的不足,这会限制模型的泛化能力;其次,模型训练通常需要比针对特定体系模型更高的计算资源。
例如基于生成对抗网络(GAN)框架开发的idpGAN模型,其训练数据来自粗粒化构象集合,但在原子级建模中的可迁移性仍然有限。GAN曾是计算机视觉领域图像生成的主流方法,但近年来扩散模型(diffusion model)逐渐成为更强大的生成框架,并在蛋白构象生成研究中得到应用。例如Tang等提出的str2str模型在PDB结构数据上进行训练,能够对部分快速折叠蛋白进行构象采样。在此基础上,Zhu等开发了IDPfold模型,该模型在IDRome结构数据上进行训练,结果表明其能够较准确地再现测试IDP体系的回转半径
需要指出的是,目前这些AI模型通常生成离散的结构构象,而无法像MD轨迹那样提供连续的构象变化信息。为了解决这一问题,研究者提出了MDgen等模型,能够为小肽体系生成连续的构象轨迹。未来随着算法的发展,这类方法有望进一步提升性能,从而直接生成连续的IDP构象轨迹,逐步弥合基于AI方法与传统MD模拟之间的差距。
表2|靶向内在无序蛋白(IDP)药物发现的部分代表性实例。
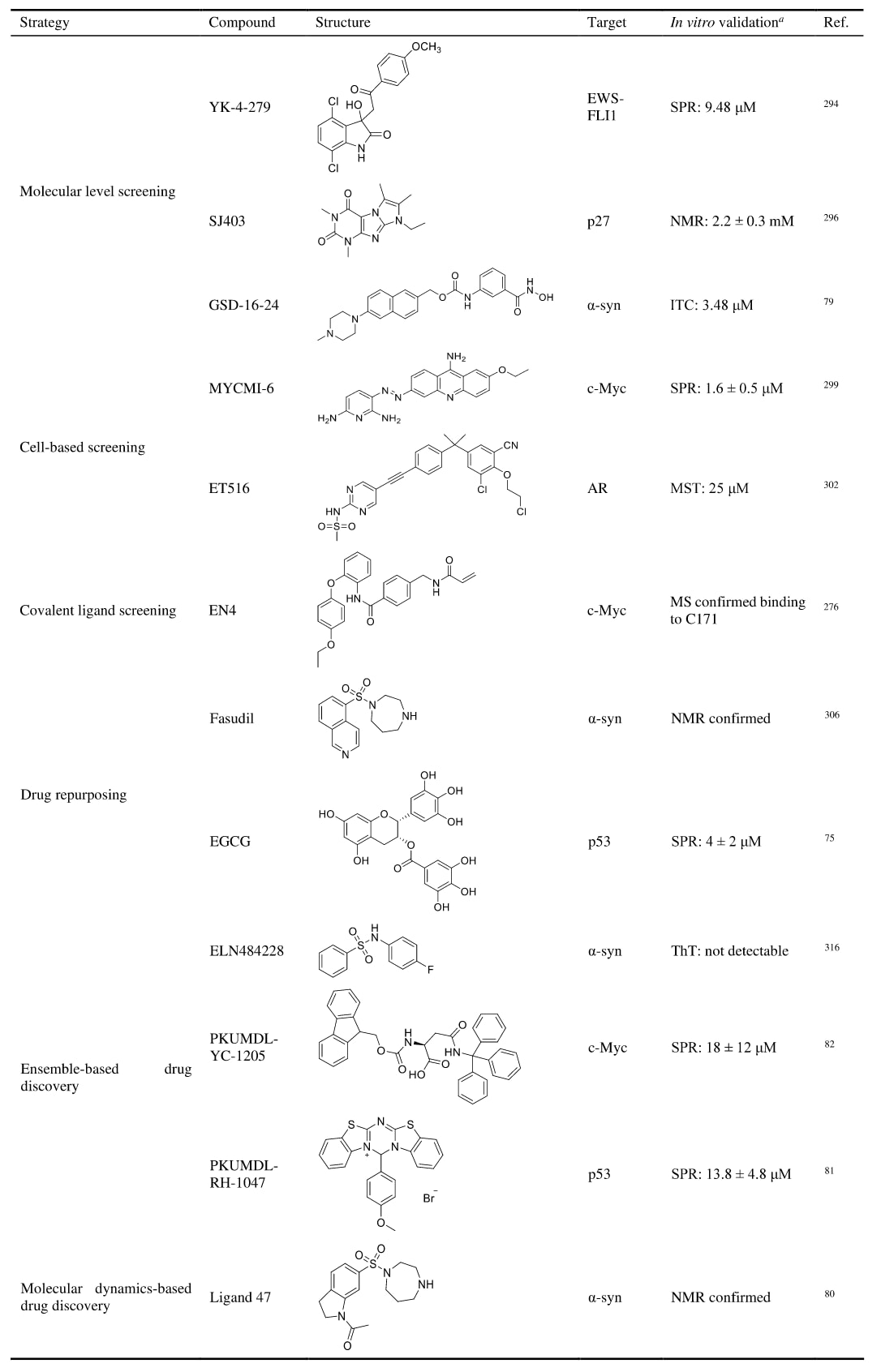
2.2.5 IDP与小分子配体的动态相互作用
当与其他分子结合时,内在无序蛋白(IDPs)可能保持无序状态,也可能转变为有序结构。部分IDP在与蛋白靶标结合时会发生结构稳定化,例如p53与MDM2结合时可形成较为刚性的结构。然而,小分子配体通常难以诱导IDP发生由无序到有序的结构转变。当前的计算与实验研究表明,IDP与小分子配体往往形成一种“模糊复合物”(fuzzy complex),其结合状态在多种不同结合模式之间持续动态转换。在这种高度动态的复合体系中,精确解析每一种具体结合模式仍然十分困难,因此相关研究通常通过平均化分析来描述整体结合特征。对已有研究结果进行综合分析后,可以总结出IDP与小分子配体相互作用的一些常见特征。
首先,IDP体系通常缺乏稳定的结合口袋。小分子配体不会诱导IDP形成固定的结合腔体,而是在由不同氨基酸残基组成的多个结合位点之间不断转移。这一现象可能源于小分子提供的焓贡献不足以补偿形成稳定结构所需的熵损失,因此有效的IDP配体往往需要更大的分子量或更高的配体效率。
其次,小分子配体在结合过程中通常偏好疏水和芳香族残基。由于IDP缺乏足够的疏水残基来形成稳定的疏水核心,小分子配体通过与这些残基发生相互作用,能够在一定程度上促进结构稳定。这一点也体现在许多IDP配体通常具有较强疏水性,并且其结合会使IDP的构象集合更加紧凑。此外,具有较高二级结构倾向的区域更容易形成包含疏水和芳香族残基的结合环境,因此小分子配体通常更倾向于与IDP中相对有序的区域结合。
第三,IDP与小分子的结合具有明显的序列特异性,在某些情况下还会涉及对特定残基的共价反应。对于共价配体而言,其非共价结合形式可以首先将共价反应基团定位在特定残基附近,从而实现残基特异性。然而,对于非共价配体而言,其序列特异性的分子机制仍有待进一步阐明。
最后,IDP配体对分子取代基变化通常非常敏感。在一些情况下,小分子中单一取代基的改变即可显著影响其活性。例如在c-Myc配体MYCi361中,仅改变咪唑环上甲基取代的位置,就可能显著削弱其对c-Myc/Max/DNA复合物的结合能力。这种高度敏感的结构-活性关系表明IDP对小分子具有较高的选择性,但其具体机制仍有待深入研究。
2.3 IDP药物设计
如图3所示,随着对IDP序列—构象集合—功能关系理解的不断加深以及新型药物设计策略的不断出现,靶向IDP的药物开发正逐渐成为现实。相关研究的发展过程通常经历多个阶段:从最初意外发现某一靶标属于IDP,到配体的发现与优化,最终推动候选药物进入临床研究。
2.3.1 当靶标被发现为IDP时的策略
在实验研究、生物信息学分析或文献调研中发现某一潜在靶基因后,其编码蛋白有时会被证明属于IDP。针对无序功能区域设计直接调控分子虽然具有挑战性,但并非不可实现。同时,多种间接调控IDP功能的策略也已被证明具有可行性。以下策略既可用于绕过IDP结构高度动态和异质性的困难,也可为直接靶向IDP做好准备。
2.3.1.1 间接靶向IDP的策略
2.3.1.1.1 基于核酸的调控策略
目前已经发展出通过调控IDP转录或翻译过程来调节其表达水平的方法,其中小分子可通过结合相应DNA或RNA实现调控。例如MYC基因中含有一个富含嘌呤的核酸酶高敏感元件III1(NHE III1),当该区域形成G-四链体(G4)结构时会抑制MYC转录。因此,稳定G4结构可以降低c-Myc的活性。目前已经报道了多种G4配体。2022年Li等对G4配体IZCZ-3进行了优化,获得更高活性的化合物,并利用其抑制MYC转录、诱导细胞死亡并抑制肿瘤生长。
编码α-突触核蛋白(α-syn)的SNCA mRNA中含有一个铁响应元件(iron-responsive element,IRE),该结构通过与铁调控蛋白结合降低翻译机器的可及性,从而抑制SNCA mRNA的翻译。Tong等发现了一种小分子Synucleozid-2.0,其能够稳定IRE结构并阻止核糖体装载,从而减少α-syn聚集诱导的细胞毒性。此外,Synucleozid-2.0还被连接到核糖核酸酶配体上形成核糖核酸酶靶向嵌合体(RIBOTAC),从而实现对SNCA mRNA的降解。
核酸结合配体能够直接降低IDP蛋白水平,因此为调控难以直接靶向的蛋白提供了一种有前景的策略。但需要注意的是,目标核酸结构往往并非某一特定基因或RNA所独有,因此在设计相关配体时需要特别关注选择性问题。
2.3.1.1.2 基于折叠伙伴蛋白的调控策略
IDP发挥功能通常依赖于与其他蛋白的结合,因此可以通过靶向这些具有稳定结构的相互作用蛋白来间接调控IDP功能。IDP通过短线性基序(short linear motifs,SLiMs)与蛋白配体结合时通常具有较低亲和力,因此可以利用蛋白质—蛋白质相互作用(PPI)抑制剂进行竞争性干扰。当IDP与蛋白配体形成复合物时,其构象集合会发生变化,在某些情况下甚至会通过无序向有序的转变形成稳定复合物,这为PPI抑制剂设计提供了结构基础。
一个经典例子是通过MDM2配体破坏p53与MDM2之间的相互作用。当无序的p53-TAD1与MDM2结合时,第19至26位残基会形成紧凑的α螺旋结构并插入MDM2的疏水口袋,从而形成稳定复合物。基于这一明确的结合模式,研究者可以针对MDM2口袋进行药物筛选,并设计能够模拟p53-TAD1相互作用基序的配体。关于PPI抑制剂设计的详细内容将在第4节进行介绍。
此外,多数翻译后修饰(post-translational modifications,PTMs)发生在IDR区域,这些修饰在调控IDP稳定性和功能方面发挥关键作用。因此,靶向参与或调控这些修饰过程的折叠蛋白也是一种有效的间接调控策略。例如Pin1是一种肽基脯氨酰顺反异构酶,其作用取决于c-Myc的磷酸化状态,可以促进c-Myc转录激活或诱导其降解。2021年Dubiella等利用小分子Sulfopin共价结合Pin1的折叠口袋,从而下调c-Myc靶基因表达。
2.3.1.2 直接靶向IDP的前期准备
根据2.2.5节对IDP—配体结合特征的分析,在尝试直接靶向IDP之前可以采取以下准备步骤。首先,预测整个蛋白序列的无序程度。IDP中存在的折叠结构域可用于设计PROTAC降解策略,而在游离状态下具有较高二级结构比例的区域通常更容易被小分子配体靶向。其次,预测序列中潜在的短线性基序(SLiMs)。具有功能性的SLiMs,特别是富含芳香族残基的区域,更适合作为潜在靶点。再次,识别能够与配体形成共价键的残基,例如半胱氨酸,因为共价配体往往具有较好的选择性和活性。此外,可以通过MD模拟评估不同IDP区域的构象特性,那些构象集合更容易受到调控的区域通常更适合作为靶点。最后,根据所研究IDP靶标的序列—构象集合—功能关系建立合适的实验体系,以支持后续药物设计研究。
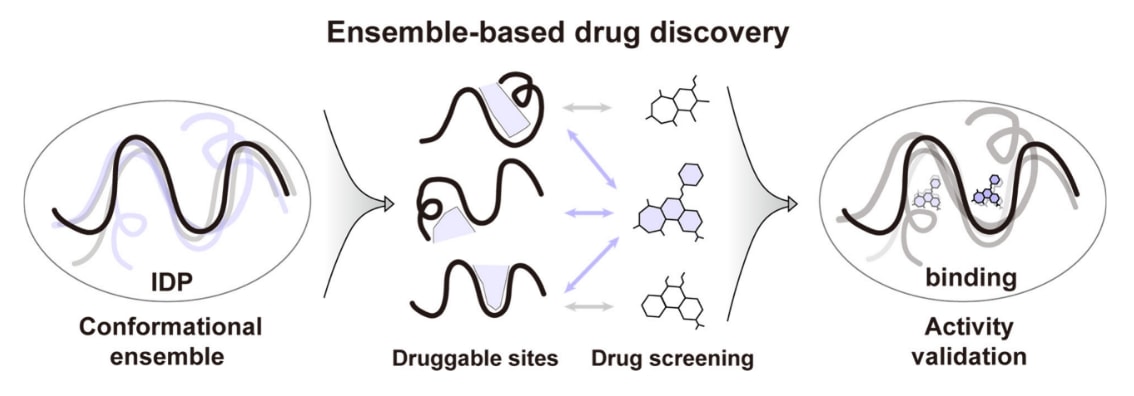
图5|靶向内在无序蛋白的基于构象集合的药物发现(EBDD)示意图。 EBDD首先从构象集合中识别出具有较高出现比例并包含潜在结合口袋的构象,作为可药物化构象;随后利用这些构象对化合物库进行虚拟筛选,并根据化合物在所有构象上的对接评分筛选候选化合物。通过EBDD发现的命中化合物通常以动态相互作用方式与IDP靶标结合。
2.3.2 如何发现能够直接靶向IDP的配体
基于核酸的调控方法在调控特定结构域功能方面缺乏精确性,而通过靶向折叠伙伴蛋白进行调控则可能因干扰该蛋白自身功能而产生较大副作用。因此,设计能够直接结合并调控IDP的配体成为解决这些问题的重要策略。对于尚未发现配体的IDP靶标,可以通过多种药物发现方法寻找能够直接结合IDP的化合物,其中计算辅助方法表现出较大潜力。
2.3.2.1 实验方法
2.3.2.1.1 分子水平筛选
直接检测化合物与IDP靶标的结合能力是最直接的策略。例如在表面等离子体共振(surface plasmon resonance,SPR)实验中,IDP被固定在芯片表面,化合物作为流动相经过芯片。当配体与IDP结合时,信号会随结合配体的质量增加而变化,从而能够检测与低丰度构象结合的配体。Erkizan等利用SPR筛选约3000个化合物,发现了能够直接结合EWS-FLI1的配体YK-4-279。
核磁共振(NMR)实验能够在溶液状态下提供IDP异质构象集合的信息,并可检测配体结合引起的构象变化。Iconaru等利用NMR筛选片段化合物库,发现了能够结合p27Kip1的配体SJ403。结合NMR和序列突变实验表明,SJ403主要结合于p27Kip1中部分形成二级结构的区域,并对芳香族残基具有明显偏好,显示出一定结合特异性。
荧光方法例如荧光相关光谱(fluorescence correlation spectroscopy,FCS)也可以敏感检测IDP与配体的结合信号,适用于IDP配体筛选,但需要注意荧光标记可能对IDP构象产生影响。此外,还可以通过基于IDP-蛋白相互作用的竞争实验筛选IDP配体。例如Zhang等利用ELISA高通量筛选发现能够破坏α-syn与LAG3相互作用的抑制剂,进一步优化得到先导化合物GSD16-24,该化合物可直接结合α-syn单体和纤维的C端区域。
2.3.2.1.2 基于细胞的筛选
根据IDP功能机制建立细胞模型也是筛选化合物的重要策略。对于已知存在相互作用蛋白的IDP,可以构建细胞内PPI抑制的高通量筛选体系。例如c-Myc与Max形成异源二聚体后才能结合DNA并发挥转录功能,因此破坏Myc-Max相互作用可以抑制c-Myc活性。Castell等将YFP分裂为两个片段分别融合到c-Myc和Max上,当Myc-Max在细胞内形成二聚体时,YFP恢复完整结构并产生荧光信号。利用该双分子荧光互补(bimolecular fluorescence complementation,BiFC)体系筛选1990个化合物,发现三个Myc-Max抑制剂。其中最强的抑制剂MYCMI-6与c-Myc的bHLHZip结构域结合,其
荧光素酶报告系统也常用于转录因子调控配体的筛选。通过将目标转录因子的转录激活结构域(TAD)与控制荧光素酶表达的DNA结合结构域融合,可以在细胞中建立筛选能够结合无序TAD区域配体的实验体系,该方法已被广泛用于AR-TAD配体的发现。
IDP在无膜细胞器形成过程中发挥重要作用,因此也发展出了基于细胞内液-液相分离(LLPS)的药物筛选方法。例如无序的AR-TAD结构域是AR凝聚体形成的主要驱动力,并与突变AR导致的耐药相关。Xie等通过LLPS筛选发现AR凝聚体抑制剂ET516。ET516直接结合AR-TAD(
2.3.2.1.3 共价配体筛选
共价配体可以与半胱氨酸、丝氨酸、酪氨酸或赖氨酸等残基形成共价键,相比非共价配体具有更持久的抑制效果。由于小分子与IDP的结合通常较弱且稳定性较低,共价配体可能在亲和力和选择性方面更具优势。不可逆结合还允许利用基于质谱的蛋白质组学方法如ABPP识别潜在脱靶位点。Boike等筛选了98种半胱氨酸反应性共价配体,发现EN4能够通过Michael加成与c-Myc的Cys171形成共价结合,从而下调c-Myc靶基因并抑制肿瘤生长。isoTOP-ABPP实验表明EN4在1400多个肽段中仅与8种其他蛋白发生脱靶作用,显示出较好的选择性。
2.3.2.1.4 药物再利用
在药物从发现到上市的过程中,人体安全性验证是一项重要挑战。药物再利用策略通过探索已知药物在新靶标或新疾病中的应用,可以显著加快药物开发进程。例如Tatenhorst等将用于治疗脑血管痉挛的Rho激酶抑制剂Fasudil重新用于抑制α-syn聚集,其通过与α-syn C端区域结合减轻小鼠模型中的病理变化。
天然产物EGCG是绿茶中的主要儿茶素,已被证明能够与多种IDP相互作用。SPR和NMR实验表明EGCG可以结合p53-TAD,并诱导其形成更加紧凑的构象,从而破坏p53-MDM2相互作用。EGCG还可结合c-Myc MBII区域并破坏其与蛋白伙伴的相互作用,同时还被报道能够结合β淀粉样蛋白寡聚体和Tau纤维。但EGCG属于具有广泛结合能力的配体,其IDP相互作用可能具有非特异性,需要进一步验证。
2.3.2.1.5 大分子配体筛选
大分子配体能够与IDP形成更多相互作用,因此可能具有更强的结合能力和更高特异性。其中核酸适配体是一类具有潜力的大分子配体。Li等筛选苏糖核酸(TNA)序列库并获得多个Myc/Max高亲和力配体,随后将其与E-box DNA元件和CRBN配体Pomalidomide连接,得到双功能分子TEP,能够以约53nM的浓度降解c-Myc。TEP与Palbociclib联合使用能够显著降低小鼠肿瘤体积。
Wang等通过适配体库筛选得到选择性结合c-Myc的适配体,并将其与Pomalidomide-PEG4-COOH连接得到双功能分子ProMyc,可在100nM浓度下诱导c-Myc降解。通过对ProMyc进行环化并加入抗PD-L1适配体提高细胞进入能力后,该分子能够显著抑制肿瘤生长。此外,含有多样结构的肽和环肽库也被认为是寻找IDP配体的有前景工具。例如Li等通过双环肽库筛选获得多个c-Myc结合肽,其中一种在高纳摩尔水平即可抑制MYC转录激活和细胞增殖。
2.3.2.2 计算方法
2.3.2.2.1 基于构象集合的药物发现
由于IDP构象集合高度异质,单一结构无法代表整个体系,因此在虚拟筛选中不应仅使用单一构象评估配体结合能力。理想情况下应评估化合物在整个构象集合中的结合亲和力,但当前计算能力难以对每个配体进行长时间MD模拟。基于构象集合的药物发现(ensemble-based drug discovery,EBDD)通过选择多个具有药物可结合特征的构象来代表整体集合,从而解决这一问题。所谓“可药物化构象”是指在构象集合中占比较高并能够形成较低能量复合物的结构。
EBDD通常包括四个步骤(图5):首先获得IDP构象集合,通常通过MD模拟结合实验数据得到;其次筛选可药物化构象并识别潜在结合口袋,可利用片段对接或CAVITY程序完成;随后将化合物库对接到这些构象上进行评分;最后根据化合物在多个构象上的结合评分评估其总体亲和力。理想候选化合物应在多个构象上具有较高评分,说明其能够适应多种构象而非仅结合单一结构。
EBDD已经成功用于发现直接结合IDP的配体。例如Tóth等利用NMR和计算方法获得α-syn构象集合并筛选出8种紧凑可药物化构象,随后对约3.3万个化合物进行对接筛选并获得候选化合物ELN484217和ELN484228。Yu等利用NMR和REMD获得c-Myc370−409的构象集合并识别三个结合口袋,从SPECS和DCSD化合物库中筛选得到6个活性化合物。其中PKUMDL-YC-1205能够破坏Myc-Max相互作用并抑制癌细胞生长。Ruan等利用MD模拟获得p53-TAD1构象集合并筛选约2万个化合物,最终得到10个结合分子,其中PKUMDL-RH-1047以
2.3.2.2.2 基于MD的药物发现
EBDD的一个不足在于无法考虑配体诱导的IDP构象变化,而MD模拟可以解决这一问题。尽管此类方法目前计算成本较高,但能够更准确地预测配体亲和力。例如Robustelli等对α-syn与Fasudil复合物进行了1.5ms的MD模拟,发现关键结合因素包括芳香堆积和电荷相互作用。随后对49个Fasudil类似物进行60μs模拟并预测其结合能力,实验NMR结果与预测一致,其中一个化合物表现出更强结合能力。
MD模拟还可用于识别IDP序列中的潜在靶向区域。例如Lama等将c-Myc划分为多个50个残基片段并进行混合溶剂MD模拟,发现MBII区域更容易在配体诱导下从延展构象转变为紧凑构象。进一步研究表明EGCG能够诱导c-Myc形成紧凑结构并破坏MBII相关蛋白相互作用。
2.3.2.2.3 IDP结合蛋白的设计
IDP在与天然蛋白配体结合时常通过无序向有序转变实现高特异性结合,因此人工设计的蛋白结合体有望成为调控IDP的重要工具。通过模仿IDP自身或其天然结合蛋白设计结合体已被证明非常有效。例如已经进入Ⅱ期临床试验的抗Myc微型蛋白药物Omomyc就是通过模仿c-Myc的bHLHZip结构域设计得到。通过截短并突变c-Myc的bHLHZip结构域获得的Omomyc能够形成同源二聚体并与c-Myc/Max形成异源二聚体,从而竞争DNA结合并抑制c-Myc转录活性。其优异的细胞穿透能力和较长半衰期使其具备临床应用潜力。
随着蛋白设计技术的发展,计算方法也被用于设计高亲和力的IDP蛋白结合体或肽配体。例如Wu等通过设计能够与IDR主链形成双齿氢键的超螺旋结构结合体,成功获得针对ZFC3H1无序区域的结合蛋白,其
Wu等还开发了一种通用的IDR结合蛋白设计方法,通过构建包含1000种不同构象的IDR-结合体模板库来匹配目标序列,并利用ProteinMPNN和AlphaFold2进行优化。该方法成功为21种IDP设计结合体,其中20种的结合能力优于200nM。以dynorphin A为例,其结合体亲和力达到7nM,结构解析结果显示dynorphin A在结合后形成稳定的延展结构,与设计模型高度一致。
对于IDP靶标而言,蛋白配体能够诱导无序向有序转变,因此在特异性和结合强度方面往往优于小分子配体。一旦解决生物稳定性和细胞进入能力等问题,蛋白类药物有望成为治疗IDP相关疾病的重要策略之一。
图6|靶向内在无序蛋白的配体优化代表性示例。 (A)c-Myc配体MYCi975在抗细胞增殖活性方面的构效关系研究。IC50值在PC-3细胞系中测定。(B)配体EPI-002对雄激素受体(AR)转录激活抑制能力的构效关系研究。IC50值通过雄激素诱导的PSA荧光素酶报告实验测定。(C)利用分子动力学模拟对AR转录激活结构域配体的亲和力进行预测。KD值通过未结合状态与结合状态的群体比例计算得到。(D)基于配体方法发现改进的Myc-Max抑制剂。Myc/Max/DNA复合物的抑制作用通过电泳迁移率变动分析(EMSA)测定。
2.3.3 如何获得更优的直接靶向IDP的配体
在获得具有初步活性的命中化合物之后,还需要进一步优化以提高亲和力、选择性以及理化性质。目前,无论是计算方法还是实验方法,在从动态异质的IDP-配体复合物中解析明确的结合模式方面仍面临困难,这限制了基于结构的药物优化策略的应用。因此,当前IDP配体的优化主要依赖于构效关系(SAR)研究,同时也采用一些非常规策略,例如共价修饰以及PROTAC技术。分子动力学(MD)模拟在解析IDP-配体结合模式方面发挥了重要作用,显示出其在促进IDP配体高效优化方面的潜力。
2.3.3.1 实验方法
2.3.3.1.1 基于SAR的优化
构效关系研究通过合成大量命中化合物的类似物,探索化学结构与生物活性之间的关系。由于IDP结构具有高度柔性,配体结构的修饰可能导致其结合模式发生变化,但现有研究表明SAR策略仍然对IDP配体有效。如图6A所示,基于已知的c-Myc配体活性数据,Zhao等人围绕MYCi975及其类似物开展研究,发现MYCi975的C5和C6位点具有结构耐受性,可以在这些位置引入亲电基团以增强活性。SAR分析进一步表明,在系列I化合物中,R1基团与核心骨架之间的连接链长度对活性至关重要,而在系列I和II中对R2位点进行甲基取代会显著降低细胞活性。该系列的先导化合物MYCi361及其无活性的区域异构体MYCi360也证实了R2位点的重要作用(图6D),说明IDP配体的活性对结构变化同样敏感。围绕MYCi975开展的SAR研究还获得了更强效的衍生物化合物37,其对细胞增殖的抑制能力提高了5倍,并在小鼠肿瘤抑制模型中表现出更好的疗效。
Basu等人利用NMR与突变实验发现,AR-TAD中的芳香族残基是液-液相分离(LLPS)的关键决定因素,而LLPS会促进AR的转录激活。鉴于EPI-002主要与AR-TAD的芳香族残基结合并抑制LLPS,该研究提出通过调节EPI-002分子中两个芳香环之间的距离与取向来调控其活性(图6B)。随后实验验证了这一策略的有效性,并获得了亲和力增强的化合物1aa。进一步在1aa芳香环上进行取代发现,在R1和R2位置引入疏水取代基可以提高活性。最终得到的化合物1ae在抑制AR介导的LLPS方面表现出更强效果,并对AR依赖的转录激活产生约5倍更强的抑制作用。
2.3.3.1.2 共价配体设计
相关研究表明,与非共价配体相比,IDP共价配体通常表现出更高的活性,这提示在配体中引入共价反应基团(warhead)有望实现更持久的调控作用。IDP共价配体可以选择性修饰序列中的特定半胱氨酸或其他反应性残基,而且这些共价修饰即使发生在已知功能结构域之外,也能够产生抑制效果。因此,如果在非共价配体的结合区域附近发现半胱氨酸或其他反应性残基,可以尝试在配体中引入共价反应基团,并对这些共价衍生物进行SAR研究,从而获得有效的共价配体。
2.3.3.1.3 PROTAC策略
当研究目标是降解IDP靶标时,将配体改造为PROTAC是一种直接增强降解能力的策略。这一方法已经应用于AR-TAD配体。与基于折叠蛋白配体构建的PROTAC类似,基于IDP配体的PROTAC同样可以通过优化连接子以及E3连接酶配体来提升活性,相关内容在第5节中进行了详细讨论。
2.3.3.2 计算方法
2.3.3.2.1 基于MD的IDP-配体结合分析
分子动力学模拟能够从原子水平揭示动态且异质的IDP-配体复合物结构,为理解IDP-配体相互作用提供结构基础。目前MD模拟主要用于解释IDP-配体的结合行为,通过整体相互作用分析来阐明机制,尚未被广泛应用于直接指导配体设计,但基于MD的高效优化策略正在逐渐成为可能。
AR-TAD配体EPI-7386目前已进入Ⅱ期临床试验(表3)。其先导化合物EPI-002能够与AR-TAD的Tau-5结构域形成共价结合,并主要与其中的芳香族残基发生相互作用。然而,这一系列化合物的具体结合模式仍不清楚。Zhu等人对EPI-002及其更高活性的类似物EPI-7170与Tau-5结构域片段进行了长时间MD模拟,以解释EPI-7170活性更强的原因(图6C)。在未结合状态下,Tau-5的R2和R3区域分别具有约30%和10%的螺旋构象。残基对协同相互作用分析显示,EPI-002和EPI-7170通过芳香相互作用结合在R2-R3界面的疏水核心区域,并诱导Tau-5形成更多的螺旋构象。EPI-7170中的二氯取代苯环能够产生更多
2.3.3.2.2 基于配体的药物发现(LBDD)
有效的SAR研究表明,IDP配体同样具有可识别的药效团特征。因此,当获得足够数量的活性配体后,可以构建药效团模型用于配体优化或开展基于配体的药物发现。Han等人利用32个已知的Myc-Max抑制剂构建了一个五点药效团模型,并对1600万化合物的数据库进行筛选,获得了一个活性命中化合物Min9。随后通过SAR分析进一步发现了改进的化合物MYCi361和MYCi975。其中MYCi975能够结合c-Myc并在低微摩尔浓度下促进其降解,在小鼠模型中表现出显著的抗肿瘤活性且具有良好的耐受性。
2.3.4 从进入临床试验的IDP药物中可以得到哪些启示
2.3.4.1 IDP作为药物靶点的安全性证据
将IDP,尤其是转录因子,作为药物靶点的一大担忧在于它们在正常细胞中具有重要功能,因此可能引发严重副作用。令人鼓舞的是,两种靶向IDP的药物Omomyc和EPI-7386(表3)在Ⅰ期临床试验中的结果表明,直接靶向IDP是可行且安全的。
EPI-7386是一种能够直接结合AR-TAD的小分子配体,可以抑制AR及其对LBD靶向药物产生耐药的突变体的转录活性。虽然EPI-7386的结构以及详细的临床前数据尚未公开,但其类似物EPI-7170在临床前研究中表现出更强的抗肿瘤活性,并在与恩杂鲁胺联合使用时增强了对AR/AR-V7的抑制作用。在一项口服Ⅰ期临床试验中,EPI-7386在转移性去势抵抗性前列腺癌(mCRPC)患者中表现出良好的安全性,在所有评估的剂量和给药方案下均未观察到剂量限制性毒性(DLT),每日剂量最高达到1200 mg时仍具有良好耐受性。同时,该药物达到了预期的靶点暴露水平,并显示出初步的肿瘤抑制效果。在另一项Ⅰ/Ⅱ期联合治疗试验中,EPI-7386(600 mg BID)与恩杂鲁胺(160 mg QD)联合用药同样表现出良好耐受性。尽管恩杂鲁胺降低了EPI-7386的体内暴露量,但其水平仍足以产生治疗作用,并表现出初步的肿瘤抑制效果,其中前列腺特异抗原下降≥50%(PSA50)的比例为88%(14/16),PSA90为69%(11/16),PSA<0.2 ng/mL为56%(9/16)。
Omomyc是一种能够结合c-Myc bHLHZip结构域的小型蛋白配体,通过将c-Myc从DNA上隔离来发挥作用。此外,Omomyc还能与自身或Max形成转录失活的同源或异源二聚体,并占据E-box序列,从而抑制MYC靶基因的转录。一项针对晚期实体瘤患者的首次人体Ⅰ期研究显示,Omomyc具有良好的安全性和耐受性,在22名患者中仅在6.48 mg/kg剂量下观察到一次DLT。Omomyc表现出非线性药代动力学特征,在6.48 mg/kg时出现组织饱和迹象,血清半衰期约为40小时。八名患者出现疾病稳定(SD),提示具有初步疗效。转录组分析表明,与疾病进展(PD)患者相比,SD患者中多种MYC转录特征被更明显地下调。蛋白质组学分析显示,SD患者中更多MYC直接靶蛋白水平下降。这些结果表明Omomyc治疗与MYC靶基因的抑制密切相关。值得注意的是,MYC水平与临床反应(SD或PD)之间未观察到相关性,这说明MYC不仅是MYC扩增肿瘤的靶点,也可能在非MYC扩增肿瘤中作为肿瘤抑制靶点发挥作用。除此之外,如表3所示,另一种肽类配体IDP121以及小分子配体WBC100等直接靶向c-Myc的药物也正在开展临床试验。
2.3.4.2 IDP作为药物靶点的未来方向与挑战
在IDP药物设计中,最迫切的需求之一是开发能够高效优化IDP配体的方法。通常第一代IDP配体的亲和力仅处于微摩尔至毫摩尔范围。许多研究表明,即使合成大量类似物并进行SAR分析,通常也只能获得有限的活性提升,使亲和力达到高纳摩尔至低微摩尔水平,而这一水平往往仍不足以满足临床需求。获得理想IDP配体的主要障碍在于对模糊复合物中IDP-配体结合模式缺乏清晰理解。因此,开发新的实验和计算方法以解析IDP-配体复合物中的结合模式,对于指导配体优化至关重要。
由于IDP-配体复合物具有高度动态性和异质性,需要具备高空间分辨率和/或时间分辨率的实验技术来提供更精确的结合信息。近年来,一些新兴技术如单分子纳米电路和先进的NMR方法已经开始用于表征IDP-配体相互作用,未来仍需进一步推广这些新技术的应用。虽然MD模拟能够在原子层面提供关于模糊复合物的重要信息,但其计算成本较高。尽管已经发展出更高效的方法来生成apo态IDP构象集合,但要在复杂构象集合中达到原子级分辨率仍然十分困难。此外,目前的计算分析方法难以评估构象集合中每一种结合模式对整体结合亲和力的贡献,因此在为配体优化提供具体指导方面仍存在局限。能够有效指导IDP配体优化的结构基础方法仍然亟待发展。
表3|直接靶向内在无序蛋白配体的正在进行及已完成的临床试验
3 靶向蛋白质别构位点的药物设计
3.1 作为药物靶点的别构调控
别构调控是一种重要的生物学机制,在多种生命过程中发挥关键作用。在这一机制中,生物大分子的正构位点(功能位点)会因远端位点(别构位点)受到扰动而发生结构或动力学变化。别构药物通过结合非功能位点发挥作用,与传统直接靶向正构位点的药物相比,通常具有更高的选择性、更低的毒性以及更丰富的调控方式,同时还可能克服耐药突变的问题。
根据对正构配体作用的影响不同,别构调节剂通常可以分为几种类型:正向别构调节剂(positive allosteric modulators,PAMs),能够增强正构配体的作用;负向别构调节剂(negative allosteric modulators,NAMs),能够削弱正构配体的作用;以及沉默型别构调节剂或中性别构配体(silent allosteric modulators / neutral allosteric ligands,SAMs/NALs),这类分子可以占据别构位点,但不会改变正构配体的作用效果。
别构药物为现代药物开发提供了一种新的研究范式,特别是在针对那些传统上被认为“不可成药”的人类蛋白时具有重要意义。这类蛋白往往具有底物结合过强、缺乏深层结合口袋,或通过难以靶向的大面积、平坦的蛋白质−蛋白质相互作用界面发挥功能等特征。由于别构调节剂并不直接与正构配体或PPI界面竞争,而是通过结合别构位点对蛋白功能进行精细调节,因此为调控这些不可成药蛋白提供了新的可能性。
尤其值得注意的是隐藏别构位点(cryptic allosteric sites)的发现。这类位点在apo结构中通常难以观察到,往往只有在特定别构配体结合后才会在holo结构中显现。对这类位点的识别能够显著拓展潜在药物靶点的范围。KRAS就是一个典型例子。KRAS是最常见突变的癌基因之一,在多种人类癌症的发生和发展中发挥关键作用,因此长期以来被认为是重要的抗癌药物靶点。然而在早期研究中,针对KRAS的小分子药物开发屡次失败,使其一度被视为不可成药靶点。近年来,随着多种KRAS^G12C^别构抑制剂的发现,这一局面开始发生改变。2021年5月28日,美国食品药品监督管理局(FDA)批准了首个KRAS别构药物Sotorasib,用于治疗携带KRAS^G12C^突变且至少接受过一次系统性抗肿瘤治疗的成人非小细胞肺癌(NSCLC)患者,这一进展极大推动了针对KRAS的药物研究。类似通过别构调节实现药物开发的“不可成药”蛋白还包括SHP2以及蛋白酪氨酸磷酸酶PTP1B等。
3.2 别构位点的预测
别构位点是设计别构药物的基础,但目前已知的别构位点数量仍然十分有限。Allosteric Database 2023(ASD2023)中目前仅收录了2422种别构蛋白和3102个别构位点。传统实验方法通常耗时较长且具有较强的偶然性,因此发展用于预测别构位点的计算方法对于加速别构药物发现具有重要意义。目前别构位点预测方法主要可以分为几类:基于正常模态分析(NMA)、基于共进化分析、基于蛋白拓扑或网络分析,以及基于机器学习或深度学习的方法(图7)。
3.2.1 基于正常模态分析的方法
正常模态分析是一种广泛用于分析别构位点的方法,其计算速度较快,相比于分子动力学模拟所需时间更短。Panjkovich等人开发了用于预测蛋白别构位点和调控位点的在线服务器PARS,该方法通过在存在和不存在模拟别构调节剂的情况下计算蛋白的正常模态。如果某一位点在模拟别构调节剂结合前后表现出显著不同的运动特征,则该位点被预测为潜在别构位点。
Greener等人开发了AlloPred方法,该方法利用基于支持向量机(SVM)的模型对蛋白口袋进行排序。该模型结合了NMA信息和口袋结构描述符,用于模拟当调节剂位于特定口袋中时蛋白动力学的变化,从而预测潜在的别构位点。
Song等人开发了AllositePro,通过结合口袋特征和NMA信息预测蛋白别构位点。该方法使用逻辑回归模型构建口袋特征模型,并利用弹性网络模型分析别构配体结合引起的蛋白动力学变化。随后将两个模型输出的加权评分进行整合,从而判断每个口袋是否为潜在的别构口袋。
然而,上述方法主要关注别构口袋本身,而未考虑其与正构位点之间的联系。由于别构调控涉及正构位点和别构位点之间的协同作用,在预测过程中引入二者之间的相关性可能提升预测准确性。Ma等人利用粗粒化NMA模型——高斯网络模型(GNM),计算正构位点与别构位点之间的运动相关性,并发现二者之间的运动高度相关。在此基础上开发了CorrSite程序,该方法通过分析所有模态中的运动相关性来预测蛋白别构位点。该方法已被整合到CavityPlus网络服务器中,可用于分析潜在的可成药口袋、计算药效团、预测别构位点以及共价配体结合位点。
Xie等人在GNM框架下进一步分析了不同运动模态中正构位点与别构位点之间的运动相关性,并揭示了别构调控的主导运动模态。在此基础上开发了更为准确的别构位点预测方法CorrSite2.0,该方法成功识别出SARS-CoV-2 3CLpro的两个难以通过其他方法预测的别构位点。为了便于使用,CorrSite2.0已被整合进CavityPlus 2022网络服务器,用户只需输入蛋白结构(可以是单体或多聚体)即可完成别构位点预测。
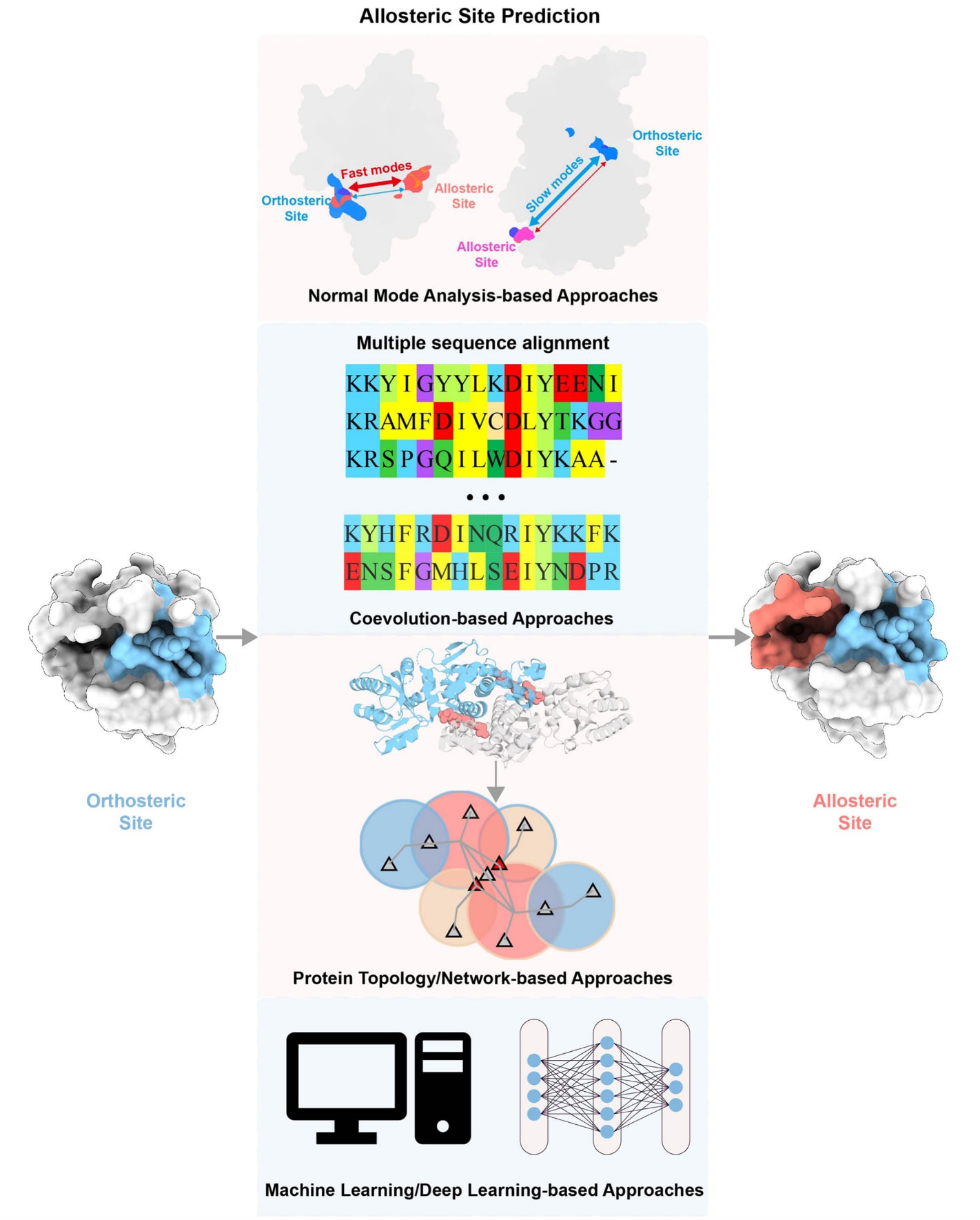
图7|别构位点预测方法。 别构位点预测方法主要可以分为四类:基于正常模态分析(NMA)的方法、基于共进化分析的方法、基于蛋白拓扑或网络分析的方法,以及基于机器学习或深度学习的方法。
3.2.2 基于共进化的方法
基于共进化的方法建立在这样一个基本假设之上:如果蛋白质中的两个位点在结构或功能上存在耦合关系,那么它们在进化过程中往往会发生协同变化。这种共进化模式可以通过多序列比对(MSA)获得,因为同源序列中记录了蛋白家族长期的进化信息。Süel等人利用基于共进化的统计耦合分析(statistical coupling analysis,SCA)方法,研究了三类蛋白家族中的氨基酸相互作用网络,包括G蛋白偶联受体、糜蛋白酶类丝氨酸蛋白酶以及血红蛋白,这些蛋白具有完全不同的折叠类型和生物学功能。研究发现,在每个蛋白家族中,残基之间的相互作用呈现出一种简单的结构模式:少量残基构成一个物理连接的网络,将正构位点与远端的别构位点连接起来。这表明,在进化过程中保守且稀疏的残基相互作用网络构成了蛋白别构通信的结构基础。
Xie等人进一步开发了KeyAlloSite方法,用于预测别构位点以及关键别构残基(allo-residues)。该方法基于进化耦合模型(evolutionary coupling model,ECM),首先通过多序列比对计算残基之间的进化耦合关系,然后分析正构位点与别构位点之间的耦合强度。研究发现,与其他非功能位点相比,别构位点与正构位点之间通常表现出更强的进化耦合关系。通过对别构口袋中残基的进化耦合值进行逐对比较,可以进一步识别关键的别构残基。KeyAlloSite能够识别那些远离催化中心但对酶催化活性产生显著影响的关键残基。相关研究还表明,即使是较弱的共进化耦合信号,也包含了重要的别构调控信息。
3.2.3 基于蛋白拓扑或网络的方法
蛋白质的整体拓扑结构在别构调控中具有重要作用,别构效应的强弱被认为在很大程度上编码于蛋白整体拓扑结构之中。Amor等人提出了一种原子水平的图论框架来揭示别构相互作用。该方法通过构建包含共价键和非共价键的原子图,并根据键能进行加权,同时定义了“键到键倾向性”(bond-to-bond propensity)指标,用于量化蛋白内部瞬时键波动在结构中的传播所产生的非局域效应。每一条键的倾向性得分反映了该键通过图结构与正构位点耦合的强度。随后通过分位数回归(quantile regression,QR)方法,将每条键与蛋白中在几何距离上与正构位点相似的一组键进行比较,从而识别显著相互作用并预测潜在的别构位点。
Wang等人开发了基于网络的分析方法Ohm,该方法仅依赖蛋白结构即可识别和表征蛋白中的别构通信网络,包括预测别构位点、别构传递路径、路径中的关键残基以及残基对之间的别构相关性。Ohm的核心是扰动传播算法。首先根据蛋白三维结构构建接触图,然后对正构位点的残基施加扰动,并计算每个残基受到扰动影响的频率,该指标被称为别构耦合强度(allosteric coupling intensity,ACI)。随后根据ACI值和残基的三维坐标对残基进行聚类,每一个聚类区域被预测为一个别构热点。在扰动传播过程中,系统还会记录扰动传播经过的残基,从而获得连接正构位点与别构位点的别构传递路径。
Xie等人还研究了蛋白拓扑结构与别构调控之间的关系,并开发了TopoAlloSite方法。该方法利用核支持向量机(kernel SVM),基于整体蛋白拓扑结构预测别构位点。首先根据蛋白三维结构在折叠层级构建蛋白拓扑图,其中每个节点代表一个蛋白结构域或别构配体。研究发现,别构位点往往位于两个或多个具有相同折叠类型的结构域之间。由于TopoAlloSite仅依赖蛋白整体拓扑结构而不依赖口袋检测,因此特别适用于预测隐藏的别构位点。
3.2.4 基于机器学习或深度学习的方法
近年来,机器学习,特别是快速发展的深度学习技术,已被广泛应用于解决多种生物学问题,例如蛋白质结构预测等。这些成功应用也推动其在别构位点预测中的应用。Demerdash等人利用支持向量机(SVM)建立预测模型,通过结构特征、动力学特征以及网络特征区分已知的别构热点与非热点位点。Huang等人则结合口袋分析算法Fpocket提供的特征,并利用SVM对蛋白口袋进行重新排序,以预测潜在的别构位点,并建立了Allosite在线服务器。
Tian等人开发了一种集成学习方法PASSer用于预测别构位点。该方法结合了极端梯度提升算法(XGBoost)和图卷积神经网络(GCNN)。其中XGBoost用于学习口袋的物理化学性质,而GCNN则用于学习口袋的拓扑结构特征。随后,Xiao等人利用两个自动化机器学习框架AutoKeras和AutoGluon开发了改进版本PASSer2.0。之后,Tian等人又提出了基于学习排序(Learning to Rank,LTR)思想的方法PASSerRank,该方法根据口袋特征与已知别构位点特征之间的匹配程度对口袋进行排序。最终,PASSer、PASSer2.0和PASSerRank被整合到PASSer在线平台中。
基于“反向别构通信”模型,即认为别构位点与正构位点之间的调控是双向的,Zha等人结合蛋白动力学、机器学习方法AdaBoost以及最短路径算法,开发了AlloReverse在线服务器,用于预测别构位点、别构残基以及调控路径。该方法还能够揭示不同别构通路之间的层级关系以及不同别构位点之间的耦合关系。
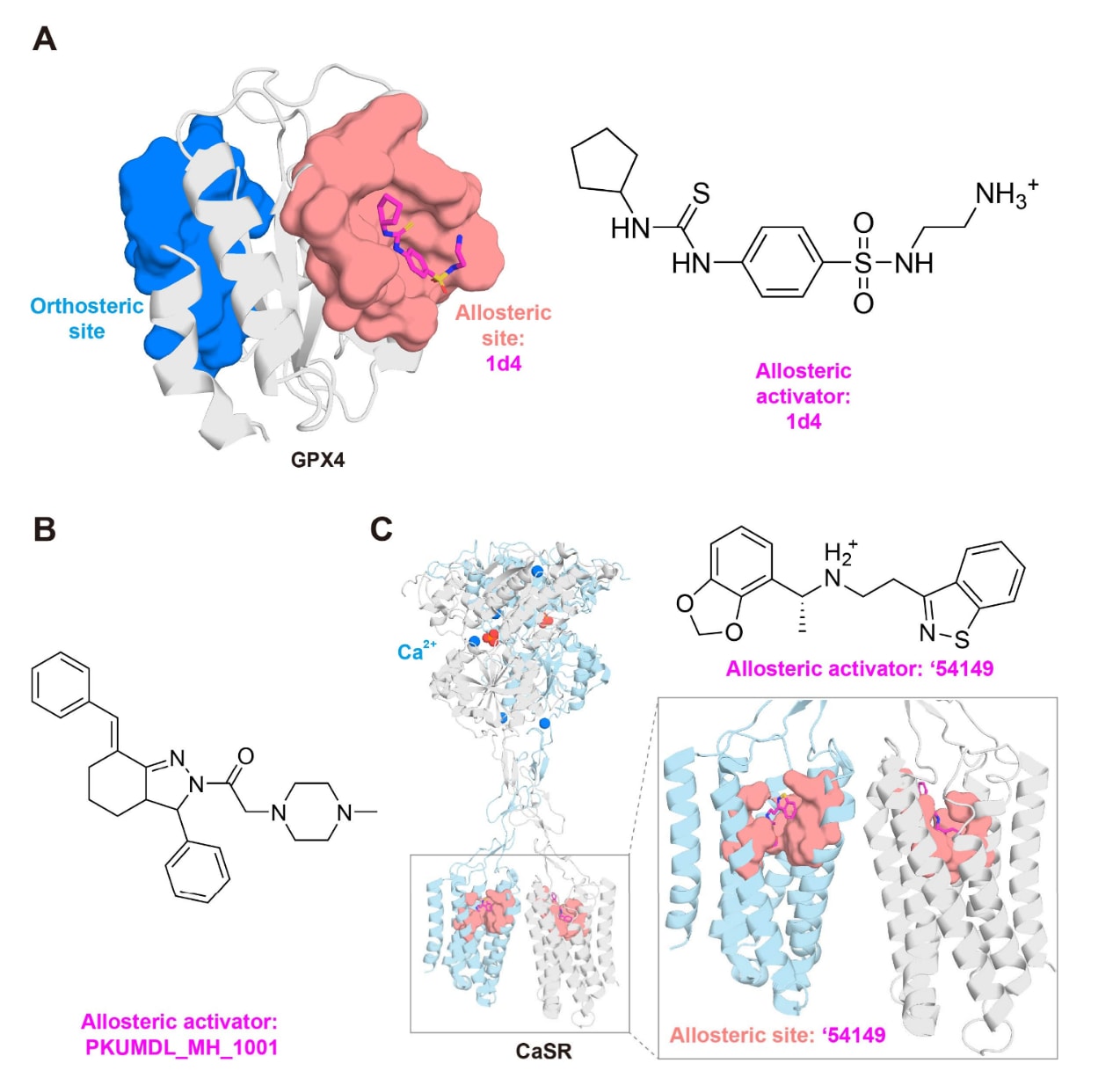
图8|蛋白别构激活剂设计的示例。 (A)GPX4与化合物1d4的对接复合物结构。(B)化合物PKUMDL_MH_1001的化学结构。(C)CaSR与‘54149复合物的晶体结构以及‘54149的化学结构,其中PO₄³⁻以红色球体表示。
3.3 隐藏别构位点的识别
3.3.1 识别隐藏别构位点的实验方法
目前已知的大多数隐藏别构位点都是通过高通量筛选以及蛋白复合物结构解析发现的。高通量筛选通常会在实验中测试包含大量化合物的化合物库(通常从数千到数百万个小分子),以寻找能够与目标蛋白结合的分子。这些分子既可能结合在已知的正构位点,也可能结合在隐藏的别构位点。当小分子结合到隐藏的别构位点时,可能诱导蛋白发生构象变化,从而稳定原本未被观察到的结合口袋。随后,通过X射线晶体学、核磁共振(NMR)以及冷冻电镜(cryo-EM)等蛋白结构解析技术,可以获得蛋白结构信息,从而确定隐藏别构位点的具体位置,并理解其形成机制。
例如,Mendes等人报道了模式生物Mycobacterium smegmatis来源的首个全长CoaBC蛋白结构,并随后对CoaB进行了高通量筛选,发现了两种具有不同化学骨架的抑制剂。通过命中化合物扩展研究,进一步获得了针对结核分枝杆菌CoaB具有高效且选择性的抑制剂。利用X射线晶体学解析的CoaB-抑制剂共晶结构表明,这些抑制剂结合在CoaB二聚体界面处的一个隐藏别构位点上。
3.3.2 识别隐藏别构位点的计算方法
由于实验方法通常耗时较长且具有一定偶然性,因此研究者开发了多种计算方法用于识别隐藏别构口袋。其中,分子动力学(MD)模拟是最常用的方法之一,在研究蛋白动力学以及识别静态晶体结构中不可见的瞬态或隐藏别构位点方面发挥了重要作用。通过模拟蛋白随时间变化的运动,MD能够揭示罕见构象以及隐藏口袋暴露的条件。
例如,Hollingsworth等人对M1、M2、M3和M4型毒蕈碱型乙酰胆碱受体(mAChRs)进行了大规模原子级MD模拟,并在M1型受体中发现了一个隐藏的别构口袋。该口袋在已有晶体结构中并不可见,但在MD模拟过程中会动态打开,从而揭示了BQZ12及相关别构调节剂的结合方式以及其亚型选择性的来源。Ilie等人通过混合溶剂MD模拟研究了不同整合素中的隐藏αI别构位点。在这种方法中,利用与水混合的小分子探针来探测蛋白中的隐藏位点。然而,隐藏别构位点何时在模拟中出现往往无法事先确定,而且这些位点通常需要较长时间尺度的MD模拟才能显现,因此计算资源消耗较大。
近年来,一些基于人工智能的方法也被用于发现隐藏别构位点。例如,Meller等人利用MD模拟数据训练了一个图神经网络模型PocketMiner,用于预测哪些口袋在MD模拟过程中更可能打开,从而识别潜在的隐藏位点。在后续研究中,通过对多序列比对(MSA)进行随机采样并使用AlphaFold生成蛋白构象集合,发现AlphaFold能够采样到完全或部分打开的隐藏口袋。这些已打开或部分打开的口袋可以作为MD模拟的初始结构,从而加速隐藏位点的发现。类似地,Vani等人开发了AlphaFold2-RAVE方法,该方法主要利用减少MSA信息的AlphaFold2生成大量潜在构象,并将其作为AI增强MD模拟的初始结构。随着人工智能技术的发展,隐藏别构位点的发现预计将越来越依赖AI驱动的方法。
3.4 别构药物设计
3.4.1 别构药物设计策略
在早期,由于对别构相互作用及其调控机制缺乏深入理解,大多数别构调节剂是通过高通量实验筛选偶然发现的。随着别构蛋白结构、别构位点以及别构调节剂相关数据的不断积累,以及计算方法的发展,逐渐提出了基于计算和基于结构的理性别构药物设计策略。理性别构药物设计通常包括几个关键步骤:识别别构位点、基于结构的虚拟筛选或de novo分子生成、实验验证以及进一步优化。
在确定别构位点之后,可以利用基于结构的虚拟筛选方法,从各种化合物库中筛选能够结合该位点的别构调节剂。也可以基于该位点直接通过de novo药物设计或深度生成模型生成新的分子。目前已经开发了多种分子对接方法,例如基于经典物理模型的对接方法Glide或DOCK,以及基于深度学习的对接方法,其中部分方法能够进行全原子柔性对接,例如DiffBindFR和AlphaFold3。这些方法均可用于基于已识别的别构位点发现潜在的别构调节剂。
近年来,还出现了一些能够直接在目标结合位点内部设计类药分子的分子生成模型。目前这些方法主要用于在蛋白正构位点生成分子,但理论上同样可以用于别构位点,尽管后者更具挑战性。通过计算设计获得的候选化合物随后需要通过实验进行功能验证,并进一步优化。
由于别构位点结合分子可能产生激活、抑制或不影响靶标活性的不同效果,在实验验证之前预测其调控类型具有重要意义。Huang等人开发了AlloType方法,利用各向异性网络模型(ANM)和线性响应理论(LRT)在理论上预测别构调控类型。通过对结合位点的力学分析发现,配体结合施加的力决定了别构调控的类型。别构耦合强度
与正构位点配体优化相比,别构分子的优化面临更大挑战,因为别构分子往往呈现平坦或浅显的构效关系,即轻微的结构修饰可能破坏活性,而且更高的结合亲和力并不一定对应更好的功能活性。在别构位点中,不同残基对别构效应的贡献并不相同。关键别构残基(allo-residues)决定了别构信号传递的方向和强度,而其他残基主要在结合过程中发挥辅助作用。因此,识别关键别构残基对于指导别构分子的优化以及理解别构调控机制至关重要。
Xie等人开发了KeyAlloSite方法,这是首个能够基于进化耦合模型(ECM)在理论上预测关键别构残基的计算方法。该方法通过多序列比对分析正构位点与别构位点之间的进化耦合关系,并通过比较别构位点中各残基的进化耦合值差异来确定关键残基。KeyAlloSite在多个典型别构蛋白(如BCR-ABL1)中的预测结果与实验研究高度一致,为别构分子的优化提供了重要工具。在进行别构分子优化时,首先预测关键别构残基,并保持或增强分子与这些残基之间的相互作用,以确保别构信号能够有效传递,同时在优化与其他残基的结合过程中获得所需活性的别构分子。
3.4.2 蛋白激活剂设计实例
3.4.2.1 GPX4别构激活剂
谷胱甘肽过氧化物酶4(GPX4)是目前已知唯一能够修复脂质过氧化物的抗氧化酶,并参与调控二十碳烷类物质的生物合成、细胞因子信号以及铁死亡。激活GPX4可以有效抑制二十碳烷类物质的产生和炎症反应,因此在炎症和铁死亡相关疾病的药物开发中具有重要潜力。然而,GPX4的别构位点此前并不清楚。
Li等人采用基于结构和计算的理性设计策略开发了首批GPX4别构激活剂。首先利用结合位点检测程序CAVITY识别GPX4表面的潜在配体结合口袋,然后利用别构位点预测方法CorrSite分析这些口袋与正构位点之间的运动相关性,从而识别潜在的别构位点。研究发现一个位于正构位点相对侧的新别构位点(图8A)。随后利用分子对接程序Glide在该位点上对SPECS化合物库进行虚拟筛选,寻找潜在的GPX4激活剂。通过酶活实验筛选得到一个GPX4激活剂,并在细胞实验中证实其能够抑制铁死亡并减少促炎脂质介质的产生。进一步通过化学合成和SAR分析对该化合物进行优化,最终获得7个额外的GPX4别构激活剂。其中最强的化合物1d4在无细胞体系中20 μM浓度下能够将GPX4活性提高至150%,在细胞提取物中的EC50为61 μM。
3.4.2.2 15-LOX别构激活剂
人网织红细胞15-脂氧合酶(15-LOX-1,简称15-LOX)是参与脂氧素(lipoxins,LXs)生成的关键酶之一。LXs是由花生四烯酸在人体白细胞中生成的一类氧化衍生物,具有抗炎作用,并能够促进炎症消退和组织稳态恢复。同时,15-LOX的产物,如15-羟基二十碳四烯酸(15-HETE)和13-羟基十八碳二烯酸(13-HODE),也具有抗炎活性。研究表明,激活15-LOX是一种调控花生四烯酸代谢网络并控制炎症的有效策略。然而,由于15-LOX的别构位点及其别构激活机制均未知,开发其别构激活剂面临很大挑战。
Meng等人提出了一种理性策略来发现15-LOX别构激活剂。首先结合互信息方法MutInf与结合口袋检测及可成药性预测方法CAVITY识别新的别构位点,然后在该位点进行虚拟筛选,并通过重组人15-LOX的无细胞实验检测筛选化合物的活性,从而发现新的别构激活剂PKUMDL_MH_1001、PKUMDL_MH_1024和PKUMDL_MH_1025(图8B)。以最强的激活剂PKUMDL_MH_1001作为分子探针,进一步研究了15-LOX激活对人多形核白细胞(PMNs)和人全血中花生四烯酸代谢网络的影响。结果表明,15-LOX别构激活剂可以通过减少促炎产物的生成并增加15-HETE的产生,从而改善炎症控制。
3.4.2.3 CaSR别构激活剂
钙感受受体(CaSR)属于C类G蛋白偶联受体(GPCR),是一种细胞表面Ca²⁺感受器,在人体钙稳态调控中发挥关键作用,也是治疗甲状旁腺疾病的重要药物靶点。CaSR以必需同源二聚体形式存在,每个亚基由一个结合Ca²⁺的胞外结构域以及一个能够激活异源三聚体G蛋白的七跨膜结构域(7TM)组成。CaSR的功能获得性突变会导致常染色体显性低钙血症1型(ADH1),而功能缺失突变则会导致家族性低尿钙高钙血症1型(FHH1)。此外,在慢性肾病中,甲状旁腺CaSR的表达和功能往往降低,从而引发继发性甲状旁腺功能亢进(SHPT)。
由于正构位点高度保守,正构抑制剂往往缺乏选择性,因此针对CaSR的治疗策略主要集中在开发靶向别构位点的激活剂或抑制剂。目前,小分子别构激活剂Cinacalcet以及肽类激活剂Etelcalcetide已被批准用于临床,但仅用于接受透析治疗的慢性肾病SHPT患者(通常为5期)。这些药物可能产生低钙血症、胃肠道问题、低血压以及无动力骨病等严重副作用。
早期晶体结构研究表明,在CaSR的活化状态下,cinacalcet和evocalcet在二聚体两个单体的7TM区域中分别呈现“伸展型”和“弯曲型”构象。基于这些结构信息,Liu等人尝试发现拓扑结构不同于已知CaSR别构激活剂的新型化合物,并采用大规模化合物库对接策略进行研究。首先利用DOCK3.7在CaSR两个7TM位点(7TMA和7TMB)上对270万分子的小型化合物库进行对接。为了探索更大规模化合物库对发现高效激活剂的影响,又利用DOCK3.8对12亿分子的化合物库在更封闭的7TMB位点上进行筛选。结果表明,与270万分子的库相比,12亿分子的库筛选能够使实验命中率提高2.7倍,并使激活剂效力提高37倍。
通过对排名靠前的对接分子进行结构优化,最终获得纳摩尔级先导化合物‘54149(EC50 = 41 nM)。随后解析了CaSR-‘54149复合物的冷冻电镜结构(图8C),发现‘54149能够促进形成一种独特的活化状态二聚体构象,该构象更接近G蛋白结合状态结构,这可能是其相较现有药物具有更高疗效和更低低钙血症风险的原因。在离体器官实验中,‘54149的效力比标准治疗药物cinacalcet高100倍,并且在小鼠体内能够降低血清甲状旁腺激素(PTH)水平,而不会引起典型的低钙血症。这项研究表明,大规模化合物库筛选在基于结构的别构药物设计中具有重要价值,并为其他复杂靶点的别构调节剂发现奠定了基础。
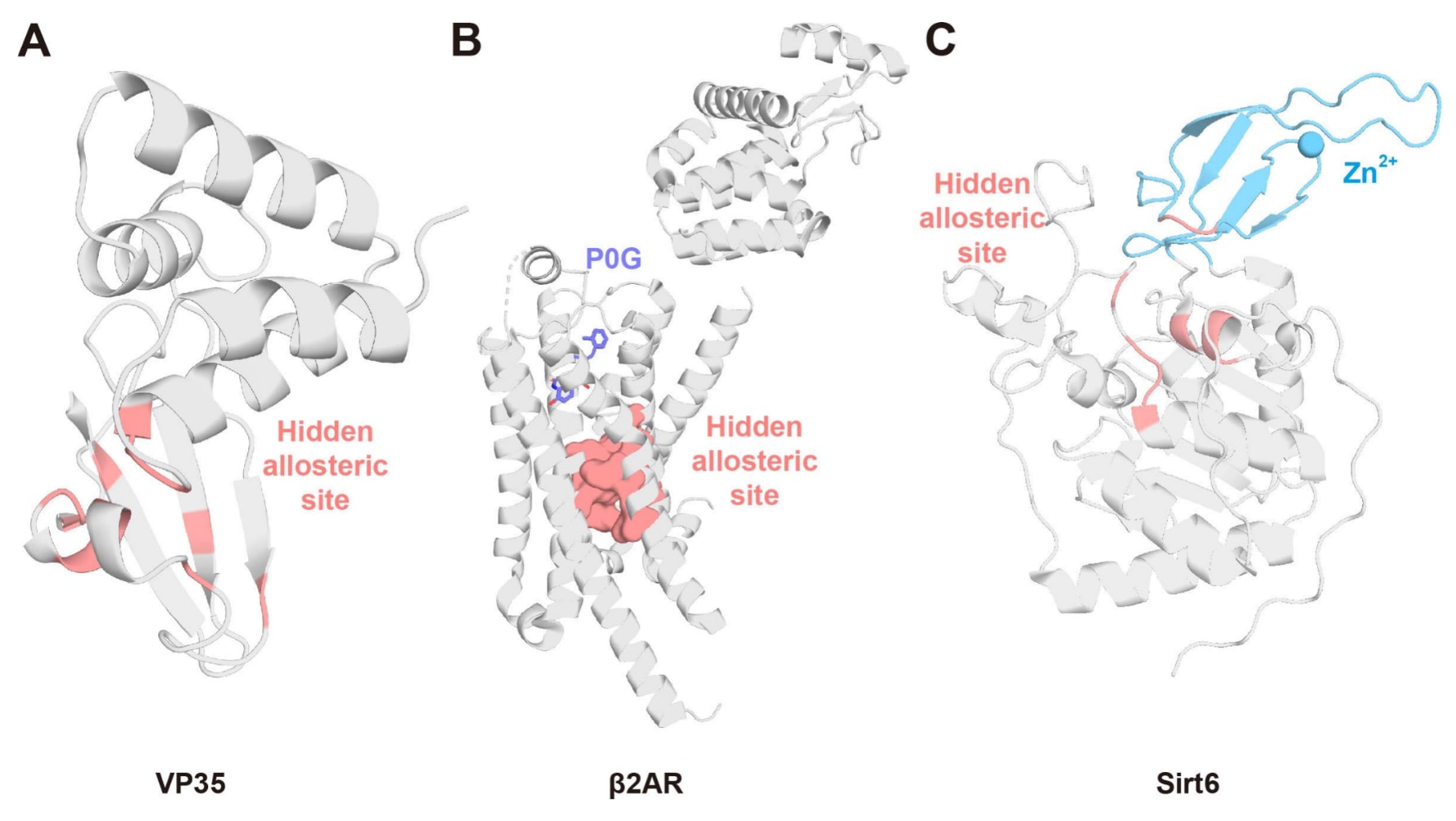
图9|靶向隐藏别构位点的药物设计示例。(A)埃博拉病毒VP35蛋白中的隐藏别构位点。(B)β₂肾上腺素能受体(β₂AR)中的隐藏别构位点,其中正构配体P0G以深蓝色棒状结构表示。(C)Sirt6蛋白中的隐藏别构位点。
3.4.3 靶向隐藏别构位点的药物设计实例
3.4.3.1 靶向埃博拉病毒VP35隐藏别构位点的药物设计
埃博拉病毒蛋白35(VP35)在病毒复制周期中发挥多种关键作用,其中一项主要功能是拮抗宿主的先天免疫反应,从而阻断干扰素(IFN)反应以及邻近细胞的信号传递,削弱抗病毒防御能力。晶体结构研究表明,VP35的干扰素抑制结构域(interferon inhibitory domain,IID)能够同时结合双链RNA(dsRNA)的末端以及其骨架结构,其中与dsRNA钝端的结合在抑制IFN反应中起主导作用。研究发现,只要降低IID与dsRNA钝端的结合亲和力,就能够缓解IFN拮抗作用并降低埃博拉病毒的致病性。因此,VP35的IID结构域成为针对埃博拉病毒及丝状病毒科其他病毒的重要药物靶点。
然而,由于VP35与dsRNA结合的界面较为平坦,小分子难以稳定结合,因此该蛋白曾被认为是不可成药靶点。Cruz等人利用特定特征波动放大(fluctuation amplification of specific traits,FAST)算法预测VP35 IID中的潜在隐藏别构口袋(图9A)。FAST是一种基于马尔可夫状态模型(MSM)的自适应采样算法,可用于探索具有特定结构特征的构象空间区域。随后利用机器学习方法DiffNets评估隐藏口袋的开启是否会影响dsRNA钝端结合界面。研究发现,隐藏口袋的开启会改变dsRNA结合界面的结构偏好,因此如果通过小分子结合稳定该开放口袋,就可能破坏dsRNA的结合。
随后通过巯基标记实验和突变实验验证了该隐藏别构口袋的存在。最终,通过模拟药物结合的共价修饰稳定了该开放构象,使VP35与dsRNA钝端的结合能力降低至少5倍。此前研究表明,只要降低约3倍的结合亲和力即可诱导宿主产生有效的免疫反应。因此,通过设计靶向这一隐藏别构位点的小分子,有望削弱病毒复制并降低其致病性。
3.4.3.2 靶向β₂肾上腺素能受体隐藏别构位点的药物设计
G蛋白偶联受体(GPCRs)是目前最大的药物靶点类别之一,约三分之一的已上市药物作用于GPCR。β₂肾上腺素能受体(β₂AR)是GPCR家族成员,在心血管和呼吸系统生理调控中发挥关键作用,也是β受体阻滞剂和β受体激动剂等常用药物的重要靶点。然而,靶向β₂AR正构位点的药物容易产生交叉反应,从而引发多种副作用。因此,开发靶向β₂AR别构位点的新型药物具有重要意义。
Chen等人开发了一种残基直观型混合机器学习框架(residue-intuitive hybrid machine learning,RHML),该方法结合了无监督聚类和可解释卷积神经网络(CNN)多分类模型。首先对β₂AR进行大规模高斯加速分子动力学模拟,以扩展采样并构建充分的构象空间。在此基础上构建RHML模型,用于确定最佳聚类数量以及别构位点开放的构象状态。随后结合FTMap以及RHML中的LIME解释器识别出一个新的隐藏别构位点(图9B)。
基于该隐藏别构位点进行虚拟筛选,最终发现一种负向别构调节剂ZINC5042。随后通过cAMP累积实验、β-arrestin募集实验以及定点突变实验对预测的别构位点及其抑制剂进行了实验验证。
3.4.3.3 靶向Sirt6隐藏别构位点的药物设计
Sirtuin 6(Sirt6)属于Sirtuin家族成员之一,该家族包含Sirt1至Sirt7。Sirt6能够催化组蛋白3赖氨酸残基的去乙酰化反应,并将乙酰基转移至NAD⁺辅因子上。Sirt6参与DNA修复、衰老等多种生物学过程,并在哺乳动物多种组织中表达,因此成为潜在的治疗靶点。然而,由于Sirt6同源蛋白之间结构高度保守,目前针对Sirt6的选择性化合物仍然较少,因此需要开发更具选择性的别构调节剂。
Ni等人通过加速分子动力学模拟并构建马尔可夫状态模型(MSM),分析了Sirt6在MD模拟过程中的关键构象状态。通过比较NAD⁺结合正构位点前后Sirt6构象集合的变化,发现了一个新的隐藏别构位点Pocket X,该位点主要由α5螺旋、连接α3和α4螺旋的环结构以及锌结合结构域(ZBD)残基组成(图9C)。Pocket X仅在新出现的M4′ MSM亚稳态中被检测到,而在holo-Sirt6的M1′至M3′亚稳态中并不存在。
随后通过定点突变实验进行验证,结果表明携带Pocket X突变的Sirt6去乙酰化活性约为野生型的一半,说明Pocket X是一个能够负向调控Sirt6去乙酰化活性的别构口袋。该位点未来可以用于设计针对Sirt6的别构调节剂。
3.5 讨论与展望
别构调控为针对不可成药靶点的药物发现提供了巨大机会。尽管目前已经开发出多种别构位点预测方法以及基于计算和结构的别构药物设计策略,但目前上市的别构药物仍只有约20种,这表明别构药物的发现仍然面临诸多挑战,并具有广阔的发展空间。
理论上,除纤维状蛋白之外的大多数蛋白都可能存在别构调控,但不同蛋白的别构调控程度差异较大,而且别构调控强度仍然难以预测。同时,别构调控类型也很难在理论上提前确定。对于酶而言,除了考虑底物结合自由能的变化,还需要分析别构调节剂是否影响催化活性,这通常需要借助量子力学计算。
此外,膜受体中的别构调控比简单的上调或下调更加复杂。例如在GPCR中,除了PAM和NAM之外,还存在偏向性别构调节剂(biased allosteric modulators)以及激动剂-PAM等多种调控形式。
在别构位点预测方面,目前大多数方法仍依赖于蛋白结构或多序列比对信息,因此对于缺乏结构信息或同源序列较少的蛋白,其预测准确性较低。未来仍需要发展能够仅基于单一序列预测别构位点、别构调控类型以及调控强度的计算方法。
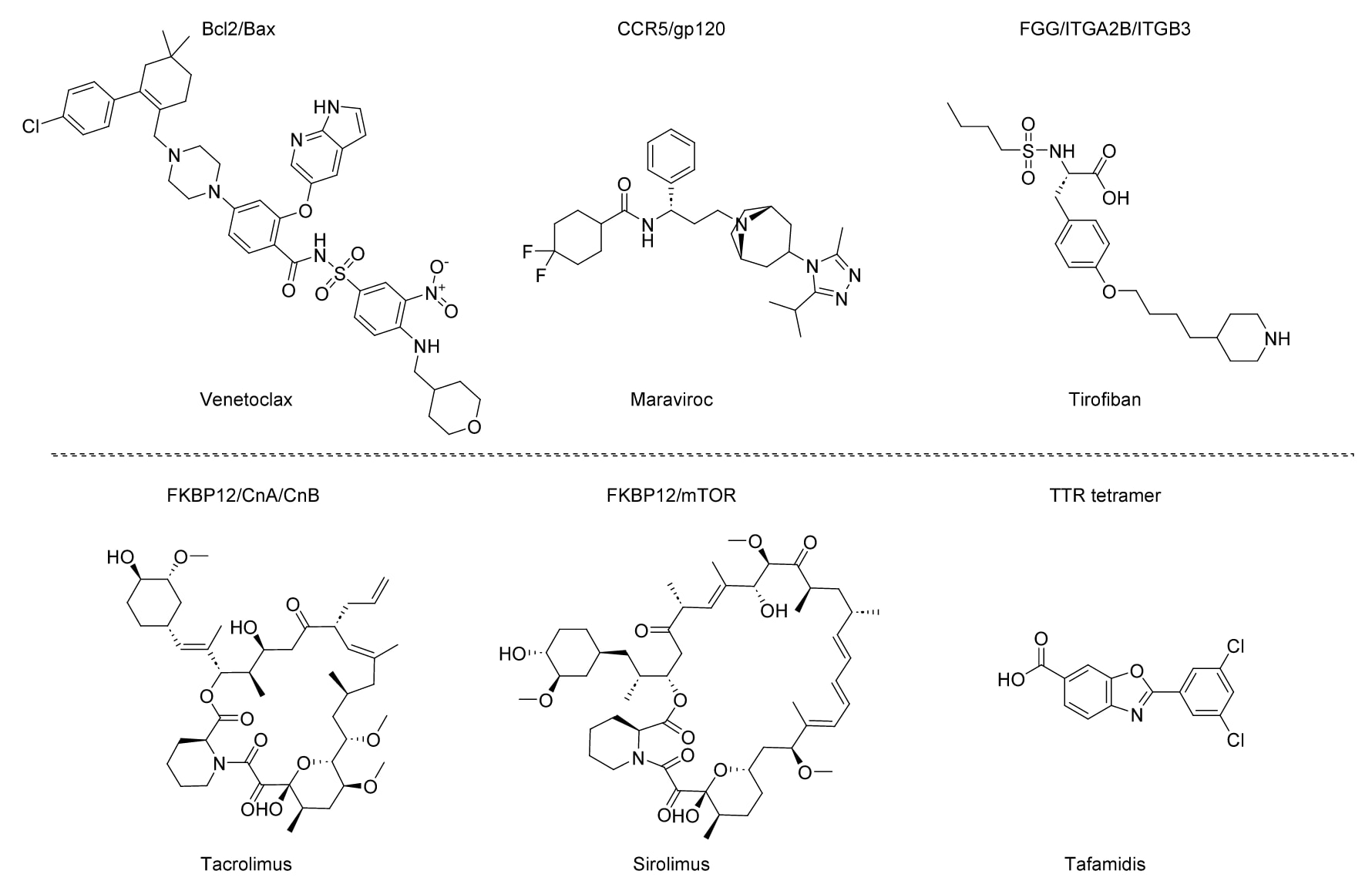
图10|六种获得FDA批准的靶向蛋白质-蛋白质相互作用的小分子药物。
4 靶向蛋白质-蛋白质相互作用的药物设计
4.1 蛋白质-蛋白质相互作用作为药物靶点
蛋白质-蛋白质相互作用(protein-protein interactions,PPIs)指两个或多个蛋白之间形成的特异性物理接触,这些相互作用通常由氢键、疏水作用以及静电相互作用等驱动。PPIs在多种生物学过程中发挥核心作用,例如细胞信号传导、细胞增殖、分化、凋亡以及侵袭等。大量研究与统计分析表明,人类数据库中大约包含19000种蛋白质,它们之间可形成约130000至650000对蛋白质相互作用。此外,PPIs在多种疾病的发生发展中也发挥关键作用。例如,一些特定的PPIs与多种癌症的进展密切相关,同时也参与阿尔茨海默病和心血管疾病的发生。因此,PPIs被认为是极具吸引力的治疗靶点,并为新型治疗策略的开发提供了巨大的潜力。
然而,由于PPIs界面通常具有平坦、面积大且高度疏水的特征,小分子药物往往难以有效靶向这些界面,因此PPIs长期以来被认为是不可成药或难以成药的靶点。首先,PPIs界面的面积通常在1500至3000 Ų之间,远大于典型小分子与靶标之间约300至1000 Ų的接触面积。其次,这些界面通常具有较高的疏水性,其核心区域通常由芳香族残基构成,并且相对于其他区域更为保守。这些核心残基使蛋白之间形成较高的结合亲和力,从而增加了小分子药物竞争结合的难度。第三,PPIs界面通常缺乏传统酶或受体靶点中常见的凹形结合口袋,而更多呈现为平坦结构,仅包含较少的沟槽或口袋,因此小分子难以实现稳定结合。此外,与传统靶点不同,PPIs通常缺乏可供参考的内源性小分子配体,这进一步增加了药物设计的难度。
在过去,针对PPIs的药物开发主要集中在抗体药物。由于抗体具有较大的结合界面以及较高的靶标特异性,已经成功应用于多个PPI靶点并实现商业化。然而,抗体药物存在生产成本高、口服生物利用度差、可能引发免疫反应以及通常无法穿透细胞膜等问题,这些因素限制了其应用范围。相比之下,肽类分子能够模拟PPI界面的复杂结构特征,并能够根据不同PPI采用多种结构形式,因此被认为是具有潜力的候选分子。与抗体相比,肽类分子的生产与修饰更为简单,但其在生理条件下稳定性较差,容易受到蛋白酶降解,体内半衰期较短且口服生物利用度较低。近年来,通过环肽设计以及引入非天然氨基酸等策略,在一定程度上提高了肽类分子的稳定性和功能表现。
此外,随着深度学习技术的发展,研究者已经能够直接根据PPI界面设计具有高亲和力的小型蛋白结合体,从而生成新的蛋白质配体。然而,这些创新设计要转化为实际应用仍需要进一步的发展和优化。近年来,小分子药物在药物开发中越来越受到重视,因为其具有明显优势,包括更好的药代动力学性质以及更强的细胞膜穿透能力,能够有效靶向细胞内PPIs。同时,小分子药物在生产、储存和运输方面也更加经济便捷。尽管小分子靶向PPIs的设计面临诸多挑战,但随着筛选技术的进步、结构解析方法的改进以及计算模拟能力的提升,小分子PPI调节剂的发现和优化取得了显著进展。目前已有6种小分子PPI调节剂(图10)获得美国食品药品监督管理局(FDA)批准,包括Venetoclax、Maraviroc、Tirofiban、Tacrolimus、Sirolimus和Tafamidis,分别用于治疗慢性淋巴细胞白血病、获得性免疫缺陷综合征、急性冠状动脉综合征、肝移植、肾移植以及转甲状腺素蛋白淀粉样变性。此外,还有超过27种靶向PPIs的小分子调节剂处于临床研究阶段。
4.2 PPI调节剂的分类
靶向PPIs的小分子调节剂根据其结合位点和功能作用可以大致分为四类:正构抑制剂、正构稳定剂、别构抑制剂以及别构稳定剂。这些不同类别具有各自的特点和研究难点,也反映出不同的发展阶段和治疗潜力。
正构抑制剂是目前研究最为广泛的一类。其作用机制是直接结合PPI界面中的某一个蛋白,从而阻断两个蛋白之间的相互作用。由于正构抑制剂的作用机制相对明确,已经发展出多种设计与优化策略,因此成为PPI调控研究的重点方向。然而,由于PPI界面通常平坦且缺乏明显结构特征,发现具有高效力和高选择性的正构抑制剂仍然具有较大挑战。
正构稳定剂同样结合在PPI界面上,但其作用方式不同。这类分子能够同时与两个蛋白发生相互作用,从而稳定或增强蛋白之间的相互作用。目前正构稳定剂的研究仍相对较少,许多相关发现往往是偶然获得。其设计难度较高,因为分子需要能够同时识别并结合两个蛋白。不过,这类分子通常不需要与内源性配体竞争强亲和力结合,并且能够结合于瞬时或特异的界面,从而提高选择性并减少脱靶效应。
别构抑制剂和别构稳定剂则通过另一种方式调控PPIs。这类分子并不直接结合在PPI界面上,而是结合于蛋白上的别构位点。通过结合这些位点,分子可以诱导蛋白发生构象变化,从而破坏或稳定PPI。因此,别构调节剂不需要直接作用于平坦的PPI界面,是调控PPIs的一种有效策略。然而,其主要挑战在于识别合适的别构位点,因为这些位点通常比正构位点更难预测。由于第3节已经对靶向别构位点的药物设计策略和挑战进行了详细讨论,该节将主要关注正构抑制剂和正构稳定剂的研究进展。
表4|用于预测热点位点的计算工具
4.3 PPI中可成药位点的识别策略
尽管PPI界面通常面积较大且看似缺乏明显结构特征,Clackson和Wells最早发现,在人生长激素与其受体的相互作用中,仅有少数残基对蛋白之间的相互作用起关键作用。这些残基被称为“热点位点”(hot spots),随后大量研究证实这一现象在PPI中普遍存在。在实验上,热点位点通常通过丙氨酸扫描突变(alanine scanning mutagenesis)进行识别。如果将某个残基突变为丙氨酸后结合自由能变化
由于热点残基对结合自由能贡献最大,因此如果小分子能够结合这些位点,就有可能显著影响蛋白之间的相互作用并调控下游功能。热点区域的面积通常约为600 Ų,仅占整个界面的约9%。此外,热点区域的理化特性使其具有较低的构象变化能垒,因此具有较高的柔性,并能够与配体形成氢键、
需要注意的是,丙氨酸扫描只能从能量贡献角度识别热点残基,而真正的可成药位点还需要具备适合小分子结合的拓扑结构。共识位点(consensus site)是指能够结合多种化学探针的蛋白区域,通常具有凹形结构并与药物结合位点重叠。研究表明,共识位点与热点位点高度相关,可视为热点的一部分。总体而言,识别热点位点是设计靶向PPI小分子的基础。当前已经开发出多种热点预测的计算方法,大体可以分为三类:基于能量的方法、基于分子动力学的方法以及基于机器学习的方法(表4)。
最早且最著名的基于能量的方法包括FOLDX和Robetta,这两种方法均用于虚拟丙氨酸扫描。其基本思路是利用侧链重排算法将界面残基逐一突变为丙氨酸,同时保持其他结构不变,然后分别计算原始蛋白和突变蛋白在结合态和未结合态下的能量变化,从而得到结合自由能的变化。FOLDX的能量函数包括范德华作用、氢键、溶剂化效应、熵变化以及库仑静电相互作用,而Robetta的能量函数则包含隐式溶剂化、Lennard-Jones相互作用、
FTMap是另一种常用的在线工具,用于识别共识位点。该方法利用经验能量函数将不同的小分子探针分子对接到蛋白表面,通过快速傅里叶变换相关算法在蛋白表面采样数十亿个可能的结合位置,并使用包含非极性包埋、成对相互作用、电荷作用以及范德华相互作用的能量函数进行评估。排名前2000的对接构象随后用于能量最小化和聚类分析,其中包含多个低能量簇的区域被认为是共识位点。此外,还有一些基于贝叶斯网络或界面分析的方法,例如PP_Site和PCRPi。大多数上述方法基于分子力学原理。近年来,Monteleone等人开发了基于量子力学片段分子轨道(fragment molecular orbital,FMO)方法的FMO-PPI,该方法能够从定点突变实验中成功识别热点,并提供蛋白-蛋白界面相互作用性质和强度的详细信息。
分子动力学模拟同样基于能量计算,并能够提供结构随时间变化的动态信息,因此可以更好地处理PPI界面的柔性并解释相互作用机制。基于分子力学/泊松-玻尔兹曼表面积(MM/PBSA)或分子力学/广义Born表面积(MM/GBSA)的自由能分解分析是一种常见策略,可用于计算单个残基对结合能的贡献并识别界面中的关键残基。此外,还可以将蛋白置于含有多种化学探针分子的溶液中,通过MD模拟探针与蛋白表面的相互作用。当系统达到平衡后,探针分子会在可结合区域聚集,从而揭示共识位点。尽管PPI界面通常较为平坦,但在小分子结合时可能发生构象变化并形成潜在结合口袋。因此,通过结合MD模拟的大规模构象采样以及传统口袋检测算法,也可以识别可成药位点。
这些方法在PPI药物设计的早期阶段被广泛应用。例如,Jiang等人利用MM-PBSA自由能分解分析研究了Nrf2与Keap1之间的关键结合残基,并发现两个重要的亚口袋。Ozdemir等人通过对Cdc42-GRD2复合物在突变前后的全原子MD模拟进行比较,分析了能量差异,从而阐明了两者相互作用的分子机制,并为后续药物设计提供了思路。
随着实验突变数据的不断积累,机器学习方法也逐渐被用于热点识别。这类方法通常利用基于序列、结构或进化信息的多种特征作为输入,并在已有热点数据上训练分类模型。由于目前可用数据量仍然有限,大多数方法采用传统机器学习模型,例如朴素贝叶斯、支持向量机、逻辑回归、决策树以及神经网络等。著名的KFC工具利用残基大小、残基类型、界面接触点以及原子接触等结构特征作为输入,并结合两个决策树模型进行预测,其召回率达到58%。随后升级的KFC2利用支持向量机模型并引入8种新的结构特征,将真正例识别率提高到0.85。其他代表性方法列于表4。值得注意的是,近期研究开始利用自动化机器学习框架AutoGluon自动选择合适模型进行热点预测。
4.4 PPI小分子调节剂的化学空间
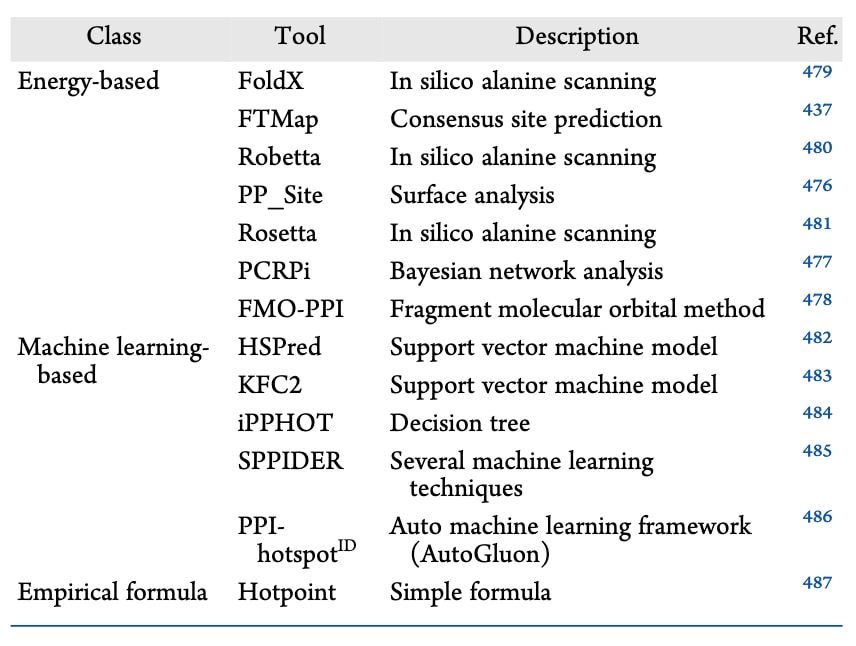
为了适应PPI界面独特的结构特征,靶向PPI的小分子调节剂在化学性质上与传统小分子药物存在显著差异。传统药物通常遵循Lipinski“五规则”,即
此外,在分子量与
此外,PPI抑制剂通常同时占据多个小口袋,这些口袋平均体积约为55 ų,而传统药物通常结合于一个平均体积约260 ų的大型口袋。对1500个PPI抑制剂的分析进一步明确了PPI抑制剂的化学空间,其典型性质包括:200 < 分子量 < 900、−1 < cLogP < 9.5、2 < 氢键受体数 < 12、0 < 氢键供体数 < 6、20 < 极性表面积 < 185、1 < 可旋转键数 < 15。
目前商业化化合物库主要针对传统药物靶点设计,其中许多分子并不位于PPI调节剂所需的化学空间内。因此,为提高PPI药物设计的成功率,有必要建立专门针对PPI调节剂的化合物库。一种简单方法是对现有化合物库进行理化性质筛选,将符合条件的分子收集到新的库中。此外,还可以利用机器学习模型区分PPI调节剂与非PPI调节剂。例如,Neugebauer等人利用分子组成、分子轮廓、官能团数量以及分子性质等描述符训练决策树模型,该模型基于25个PPI抑制剂和1137个FDA批准药物建立,在测试集上获得超过0.9的真正例识别率。
Bosc等人基于2P2IDB和iPPI-DB数据库训练了随机森林、支持向量机和决策树等多种模型,其中表现最好的支持向量机模型AUC超过0.8。随后利用这些模型从ZINC数据库中预测潜在的PPI调节剂,并经过理化性质筛选和聚类分析,建立了包含10314个分子的PPI调节剂富集化合物库Fr-PPIChem。针对免疫检查点CD47及其受体SIRPα的高通量实验表明,与普通化合物库相比,Fr-PPIChem库中活性分子的比例提高了46倍。这一研究表明,通过机器学习构建富集PPI调节剂的化合物库具有重要意义,也为未来PPI药物设计提供了有效策略。
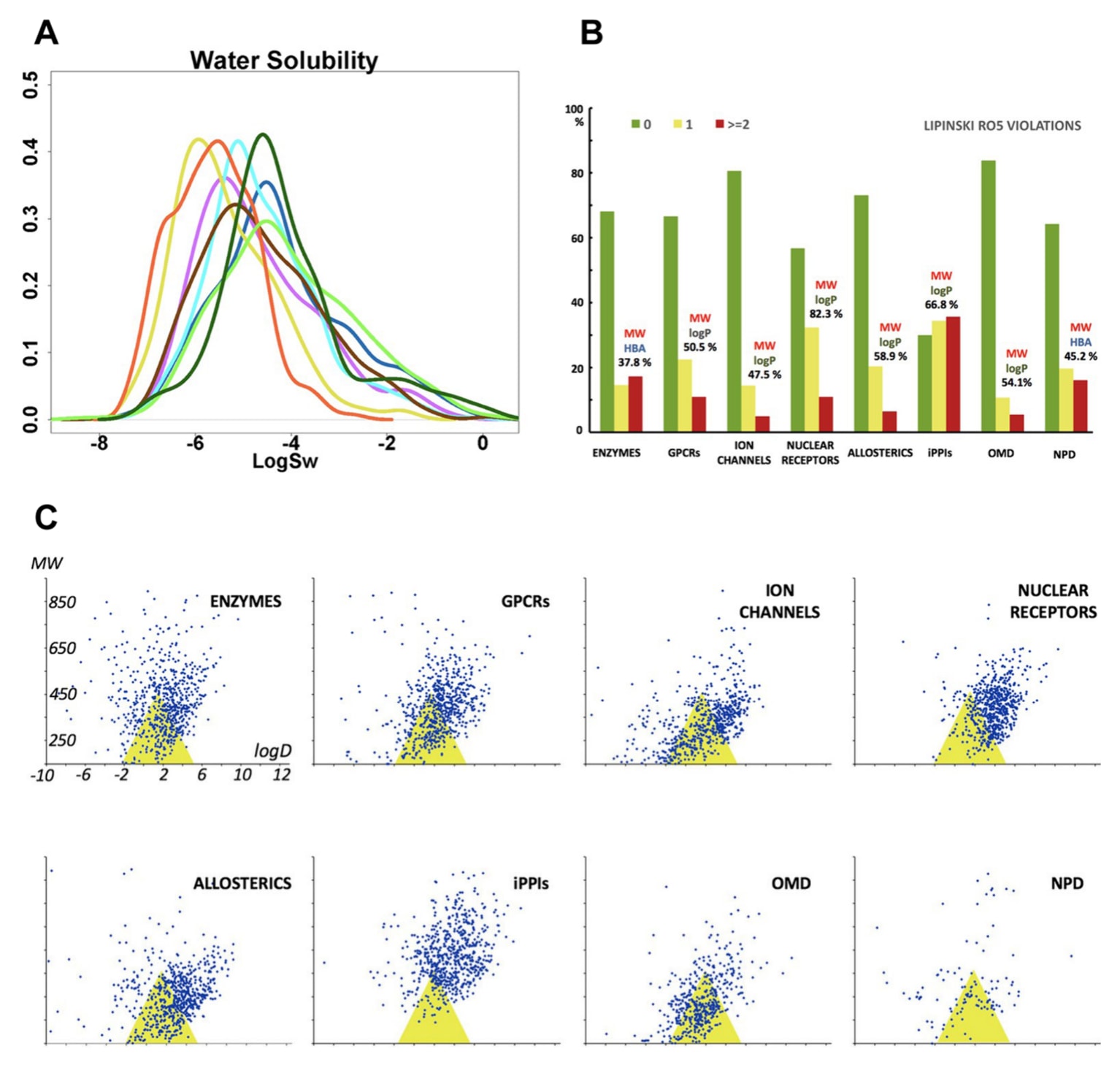
图11|靶向蛋白质-蛋白质相互作用的小分子调节剂与其他类型靶点药物分子的特征比较。 (A)溶解度预测。包括酶类(浅蓝)、离子通道(蓝色)、GPCR(紫色)、核受体(黄色)、别构调节剂(棕色)、PPI抑制剂(iPPIs,橙色)、口服上市药物(OMD,浅绿色)以及天然产物来源上市药物(NPD,深绿色)。(B)Lipinski五规则(RO5)违反数量的直方图分布。(C)各数据集在“黄金三角”中的分布示意图。位于黄金三角区域内的分子(蓝色点)通常具有更好的膜通透性以及较低的体内清除率。图转载自文献493,经许可使用。版权归David Lagorce等人所有(2017),依据Creative Commons Attribution 4.0 International License发布:http://creativecommons.org/licenses/by/4.0/。
4.5 小分子PPI调节剂的设计策略
早期针对蛋白质−蛋白质相互作用(PPI)的小分子设计主要依赖高通量实验筛选。然而,由于未充分考虑PPI调节剂所处的特殊化学空间,这类方法的命中率通常较低。随着结构生物学数据的不断积累以及“热点(hot spots)”概念的提出,基于理性设计的小分子PPI药物开发逐渐成为可行策略。一般而言,理性设计首先需要获得蛋白质复合物的三维结构,其次识别界面上的关键热点残基,随后依据这些结构信息进行分子设计或虚拟筛选。
目前用于设计小分子PPI调节剂的计算策略主要包括四类:基于查询(query)的设计策略、基于片段的策略、基于分子力学或药效团模型的策略以及基于机器学习的策略。基于查询的策略通过模拟蛋白质界面关键相互作用来构建小分子结构。由于界面热点通常呈离散分布,基于片段的策略通过片段连接、片段优化或片段自组装等方法,将多个已知低亲和力片段组合,从而获得更有效的化合物。基于分子力学或药效团模型的策略则结合分子动力学模拟、分子对接以及药效团模型,从已有化合物库中筛选潜在分子。机器学习策略则利用多种模型从已有PPI调节剂数据中学习规律,并将这些模式应用于新的化合物数据集中,以提高筛选效率和预测能力。
4.5.1 基于查询的设计策略
在该策略中,query通常指供体蛋白中对结合起关键作用的热点残基。基于查询的方法大体可分为两类:基于锚点(anchor)的设计方法以及蛋白质二级结构模拟方法。
锚点是query中的某个关键残基,该残基通常深埋于受体蛋白内部,并与相对稳定的结合口袋发生作用,因此可以作为设计的起始点。在此基础上可以采用三种扩展方式。第一种方法通过对锚点侧链进行子结构搜索以寻找新的骨架结构,然后在此骨架基础上进一步扩展,以增强与其他热点残基的相互作用,从而提高结合亲和力。第二种方法利用de novo分子设计软件或虚拟化合物库构建工具,对锚点结构进行延伸。第三种方法则通过寻找锚点的生物电子等排体(bioisosteres),以模拟其与受体蛋白之间的关键相互作用。
锚点策略已被应用于多种PPI体系的调节剂设计,其中最成功的案例之一是p53/MDM2体系。研究发现,p53 α螺旋上的三个疏水残基F19、W23和L26形成一个关键热点区域,其中W23深嵌入MDM2的凹形结合口袋中。因此研究人员以W23作为锚点进行子结构搜索,最终得到螺环氧吲哚(spirooxindole)骨架。在该结构中,吲哚环用于模拟W23侧链的结合模式,而两个疏水取代基则用于模拟F19和L26侧链的相互作用。初始获得的抑制剂对p53/MDM2相互作用的抑制常数为
在多数PPI体系中,三种关键二级结构元件——α螺旋、β转角以及β折叠——在相互作用中发挥主要作用。因此,寻找能够模拟这些结构关键相互作用的小分子,往往是一种更加直接且有效的策略。构建此类模拟分子通常有两种思路。第一种是拓扑模拟(topographical mimetics),即模拟目标二级结构中热点残基的空间取向及其组成特征。第二种方法则侧重于复制这些相互作用的生物学功能,而不必严格保持结构上的相似性。
近年来,Celis等人开发了一种基于query匹配的PPI抑制剂发现计算框架。该方法首先将热点残基映射到相应的二级结构上以构建query,然后通过比较分子形状相似性,从虚拟化合物库中筛选候选小分子(图12)。随后利用该框架设计了针对p53/MDM2和GKAP/SHANK1-PDZ体系的抑制剂。实验结果验证了该方法的有效性。例如,从p53中提取包含三个热点残基的短肽序列“ETFSDLWKLLPEN”作为query,通过FastROCS工具进行结构相似性搜索并结合分子对接筛选候选分子。最终得到的化合物A1在竞争荧光各向异性实验和
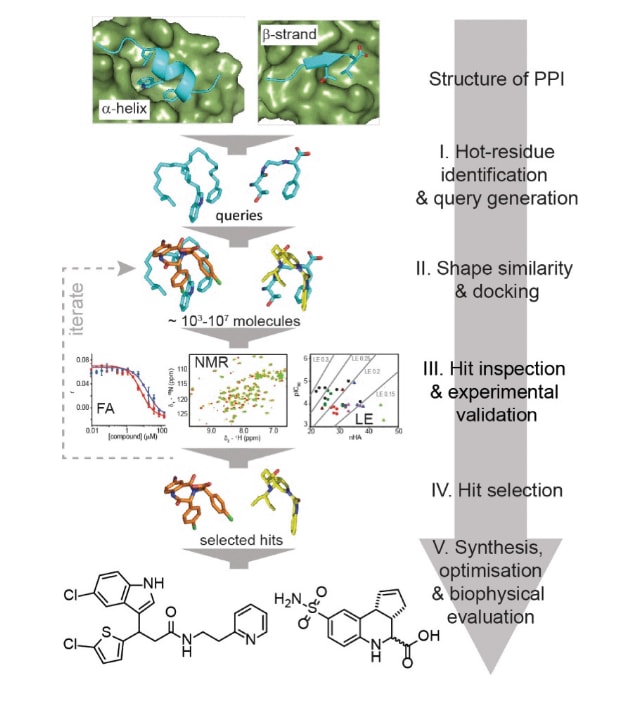
图12|展示了查询引导的蛋白质-蛋白质相互作用(PPI)抑制剂发现机制示意图,其中小分子可能通过模拟不同的二级结构来发挥作用。
4.5.2 基于片段的策略
PPI界面通常包含多个分散的小型结合口袋,而通过随机实验筛选获得的小分子往往只能结合其中一个亚口袋,从而导致结合亲和力较弱。基于片段的药物设计(fragment-based drug design,FBDD)通过将多个分子片段连接在一起,使分子能够同时占据多个口袋,从而有效提高结合能力(图13)。初始片段既可以通过实验筛选获得,也可以借助分子对接等虚拟筛选方法得到。然而,FBDD高度依赖结构信息的准确性,需要确认小分子片段确实结合在目标位点上,并确保在片段连接后分子构象不会发生显著变化。
FBDD不仅在传统药物靶点的设计中取得成功,也被广泛应用于PPI调节剂的开发,例如Bcl-xL/Bad、IL-2/IL-2Rα以及TNF-α/TNFR1抑制剂的设计。Zhao等人利用Glide程序将片段库中的分子对接到BRD4(I)结合位点,并通过X射线晶体学鉴定出9个结合片段。随后结合晶体结构提供的相互作用信息,并将候选片段的结合模式与已知高活性化合物JQ1进行比较,通过片段生长策略最终获得活性提高超过10倍的化合物。
Li等人提出了一种结合多配体同时对接(multiple ligand simultaneous docking,MLSD)和片段连接的药物再利用策略。该方法首先将多个分子片段同时对接到热点位点上,然后将命中片段连接形成模板分子,再以该模板在已获批药物库中进行相似性搜索。通过这一策略,Raloxifene和Bazedoxifene被识别为IL-6/GP130相互作用的潜在抑制剂。癌细胞实验表明,这两种化合物能够结合GP130并选择性抑制IL-6诱导的STAT3磷酸化。
基于片段的方法同样可用于PPI稳定剂的设计,其中多数策略依赖实验获得的初始片段。例如Guillory等人将接头蛋白14-3-3与致癌转录因子TAZ的复合物结构浸泡在片段混合物中,通过X射线晶体结构解析识别出能够同时与两种蛋白结合的片段,从而为PPI稳定剂的设计提供了起始结构。
4.5.3 基于分子力学与药效团的策略
分子对接、分子动力学(MD)模拟以及药效团建模是药物设计中常用的虚拟筛选技术。分子对接能够预测小分子的结合位点及其结合构象,并提供计算得到的结合能信息。对于尚未深入研究的PPI靶点,可以根据预测得到的热点残基构建药效团模型,从化合物库中筛选潜在分子。MD模拟则能够揭示蛋白质结构的动态变化,不仅可用于识别热点残基,还可对蛋白质初始构象进行广泛采样。此外,MD模拟还可用于评估小分子-蛋白质复合物的稳定性并探索相关信号机制,因此在虚拟筛选过程中具有重要价值。
例如Khanna等人采用多构象策略对uPA/uPAR体系进行虚拟筛选。研究对两个已知的uPAR晶体结构进行MD模拟,构建包含50种构象的构象集合,并利用AutoDock4将小分子对接到这些构象中。经过聚类分析后选择排名靠前的化合物进行实验验证,初步筛选得到的分子具有
Xu等人提出了一种结合分子对接和MD模拟的通用虚拟筛选框架(图14)。研究通过虚拟丙氨酸突变扫描和MM/PBSA能量分解确定热点残基,并利用基于MTDH-SND1复合物
上述抑制剂的设计通常基于单一蛋白结构进行虚拟筛选,而PPI稳定剂的设计则需要同时考虑两种蛋白之间的相互作用。一种方法是分别以PPI中两种蛋白结构进行多轮对接,然后综合比较两者的对接评分以筛选潜在稳定剂。另一种方法则直接以PPI复合物结构作为对接输入,并将结合口袋限制在两种蛋白的界面处。例如Sijbesma等人基于14-3-3与碳水化合物反应元件结合蛋白(ChREBP)的复合物结构构建结合口袋,并采用诱导契合对接方法考虑口袋中侧链构象的变化。在对471个化合物进行对接并进行人工筛选后,选择13个分子进行实验测试,其中两种化合物分别使ChREBP与14-3-3之间的结合亲和力提高约10倍和4倍。
此外,Chen等人提出了一种类似的计算流程用于设计PPI稳定剂。该流程首先利用实验或预测得到的复合物结构,在PPI界面识别潜在结合位点。可以使用如Fpocket等口袋预测算法定位可成药口袋,并结合MD模拟进一步发现界面上隐藏或瞬时出现的结合口袋。在确定合适的结合位点后,再通过分子对接和MM/GBSA自由能计算对候选分子进行筛选,从而获得潜在的PPI稳定剂。
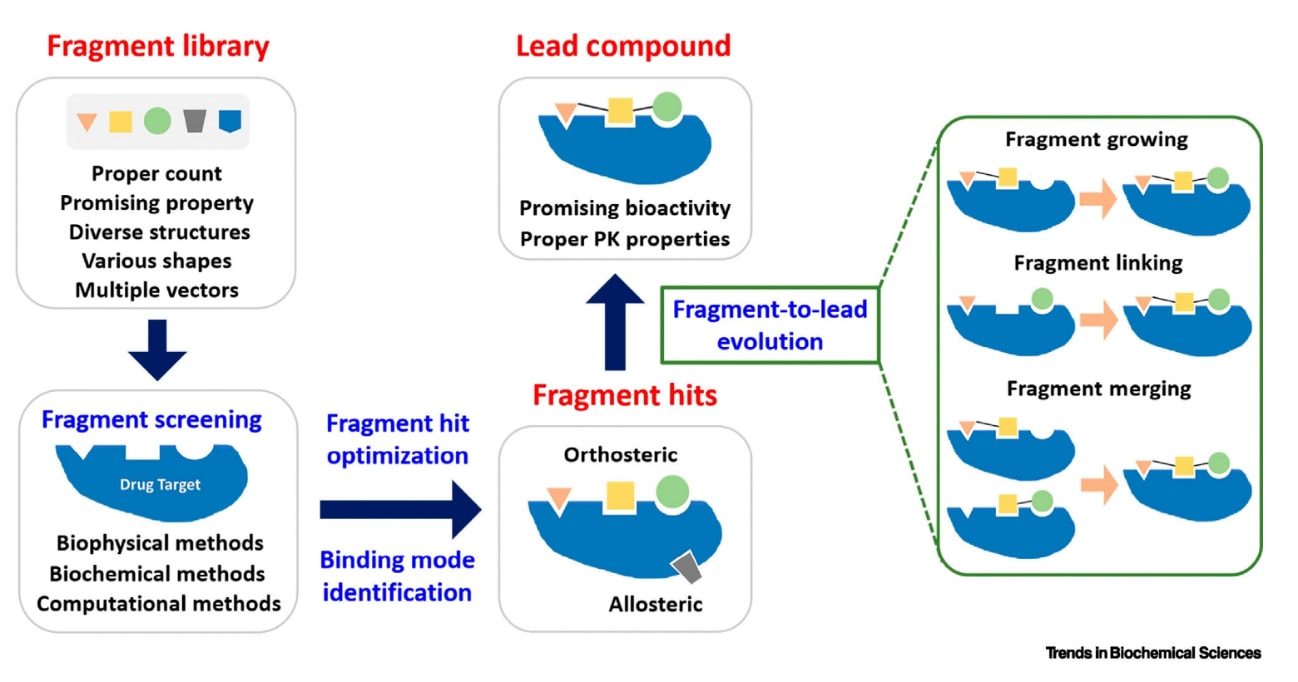
图13|基于片段的药物发现的一般流程。
4.5.4 基于机器学习的策略
随着结构表征技术和化学生物学方法的快速发展,出现了许多专门收集小分子PPI调节剂数据的数据库,例如iPPIDB、2P2IDB、TIMBAL以及DLiP-PPI library。这些数据库包含数千条小分子PPI调节剂的活性数据,为机器学习模型的训练提供了重要基础。目前数据库中涉及的PPI体系主要包括Bcl2/Bax、MDM2/p53、RBD/hACE2、Bromo/Histone、CD4/gp120、XIAP/Smac、RAS/SOS1以及Keap1/Nrf2等。
早期模型如SMMPPI、PPI-ML、pdCSM-PPI和SELPPI通常以分子指纹、拓扑特征或理化性质作为输入特征,并利用支持向量机、随机森林等传统机器学习算法建立分类或回归模型。这类模型主要用于预测某一分子是否可能成为PPI抑制剂,或估计其具体活性。近年来提出的HiGPPIM模型则引入图神经网络,用于提取更加复杂的分子图结构特征。
然而,上述模型通常高度依赖已知小分子的结构数据,因此在面对新的PPI靶点时适用性有限。更合理的策略是同时将蛋白质信息纳入模型中。例如MultiPPIMI模型同时使用分子的SMILES表示以及参与PPI的蛋白质序列作为输入。该模型通过图神经网络提取分子图特征,并利用大规模预训练蛋白语言模型ESM2从蛋白质序列中提取特征,从而提高模型的泛化能力(图15)。此外,还开发了一些针对PPI体系的三维分子生成模型,例如iPPIGAN。
现有机器学习方法几乎都集中于PPI抑制剂的预测,其分类准确率通常超过0.8。这主要是因为目前可用于训练的数据中,抑制剂数量远多于稳定剂,而稳定剂相关数据仅有数百条。在这些方法中,仅HiGPPIM模型被用于区分PPI稳定剂与PPI抑制剂,其分类准确率达到0.94。尽管这些模型表现出较好的预测能力,但现有研究在构建训练集和测试集时通常未严格控制分子相似性,这可能导致模型过拟合。此外,这些模型的预测结果仍缺乏系统性的实验验证,因此其在实际应用中的表现仍有待进一步探索。
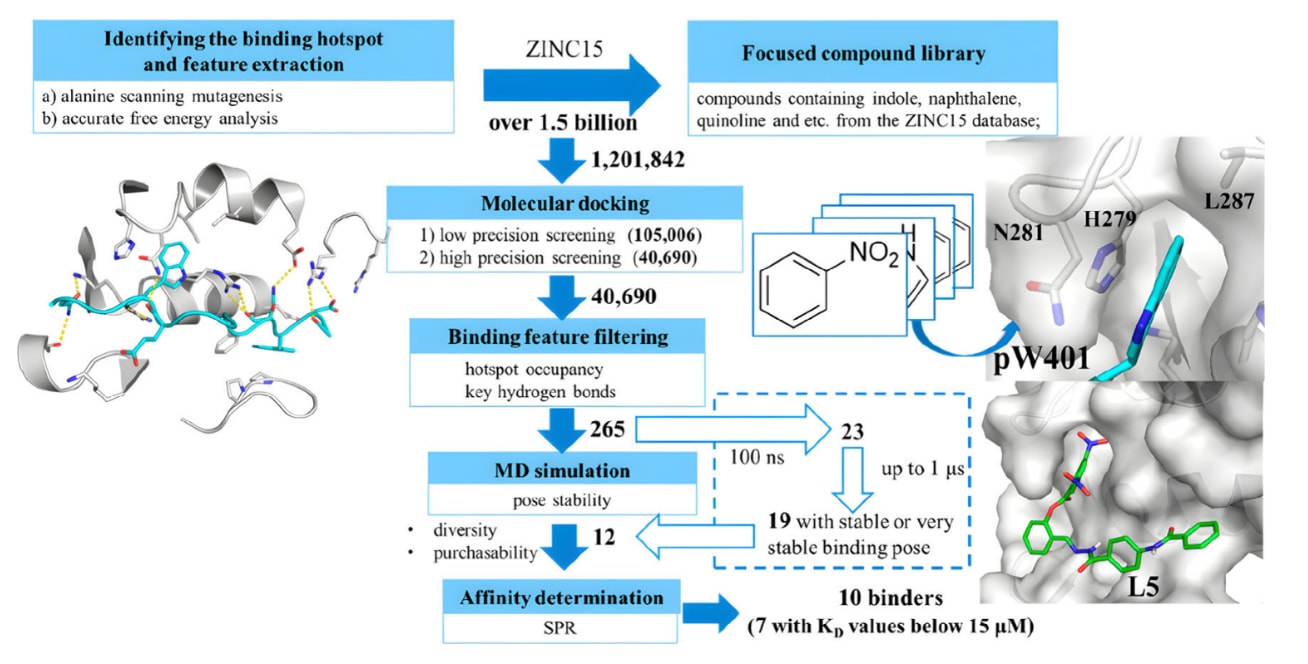
图14|结合分子对接与分子动力学(MD)模拟的通用虚拟筛选框架。
4.6 讨论与展望
蛋白质-蛋白质相互作用(PPI)调节剂的设计,核心基础在于识别结合界面上的热点残基。目前,热点预测技术已显著成熟,在诸多案例中广泛应用,为PPI药物发现奠定了坚实基础。当热点残基位置邻近且能形成合适结合位点时,可设计正构调节剂靶向这些区域;然而,若界面缺乏明确热点,或热点分布过于分散以致无法形成有效结合位点,变构调节剂往往通过诱导构象变化间接调控PPI,表现出更优效果。
正构调节剂的设计方法主要包括基于查询的策略、基于片段的策略、基于分子力学或药效团的方法,以及基于机器学习的策略。其中,基于查询的策略主要用于设计抑制剂,后三种方法则同时适用于抑制剂与稳定剂的开发。就计算手段设计稳定剂而言,相关研究仍较有限——由于缺少清晰的设计原则,目前稳定剂的发现仍主要依赖高通量筛选等实验方法。此外,稳定剂数据的匮乏也制约了机器学习方法的应用,现有模型多聚焦于抑制剂。鉴于稳定剂在由突变或功能丧失引发的疾病中具备重要治疗潜力,未来应加强对稳定剂的探索,开展更多计算尝试,并深化对其作用机制的理解。
其次,当前用于PPI调节剂的机器学习方法仍处于早期阶段,泛化能力有限,且缺乏实验验证。解决这一问题的关键在于利用实验数据对模型进行迭代优化,以提升其性能与可靠性。最后,尽管已明确PPI调节剂与传统药物分子在理化性质上存在显著差异,但针对这一独特化学空间的认知与利用仍显不足。多数研究仍使用常规商业化合物库,部分原因在于针对PPI调节剂的专用库稀缺且成本高昂。因此,未来需优先借助先进合成技术开发PPI调节剂的专用化合物库,丰富其化学多样性,进而提高筛选命中率。
5 靶向蛋白质降解的药物设计
5.1 蛋白质降解概述
蛋白质的合成与降解是生命活动中至关重要的过程。在真核细胞中,细胞内蛋白质降解的主要途径是泛素-蛋白酶体系统(ubiquitin-proteasome system,UPS)。该系统由泛素激活酶(E1)、泛素结合酶(E2)、泛素连接酶(E3)、泛素分子、26S蛋白酶体以及目标蛋白共同组成。近年来,靶向蛋白质降解(targeted protein degradation,TPD)已成为药物研发的重要方向并受到广泛关注。根据作用机制和化学结构的不同,降解剂通常可分为两类:双功能分子降解剂以及分子胶降解剂。
双功能分子降解剂的典型代表是蛋白降解靶向嵌合体(proteolysis-targeting chimera,PROTAC)。PROTAC由E3连接酶配体和目标蛋白(protein of interest,POI)配体组成,两者通过柔性或刚性的连接臂连接。该结构设计能够将目标蛋白与E3连接酶拉近,从而促进POI的泛素化并随后被蛋白酶体降解。与传统小分子抑制剂不同,PROTAC依赖“事件驱动”的降解机制,而非“占位驱动”的结合机制。这种策略显著降低了对靶点作用模式的依赖性。PROTAC只需要能够结合POI的配体,而不必是传统意义上的抑制剂,因此能够拓展对以往难以成药蛋白的调控能力。
分子胶(molecular glue)是一类能够诱导两个蛋白靠近的小分子,通过促进蛋白之间的相互作用,实现对蛋白折叠、定位或降解过程的精确调控。分子胶降解剂(molecular glue degraders,MGDs)主要通过诱导E3连接酶与底物蛋白之间形成新的蛋白−蛋白相互作用,从而实现靶向蛋白降解。与通常形成三元复合物的PROTAC不同,MGD首先与E3连接酶形成二元复合物,在此基础上形成新的蛋白-小分子界面以结合目标蛋白(图16)。这种独特机制使得MGD在不依赖靶蛋白特定结合口袋的情况下,也能够降解一些传统上难以成药的靶点。此外,由于缺少连接臂结构,MGD通常具有更小的分子量以及更有利的药代动力学性质。
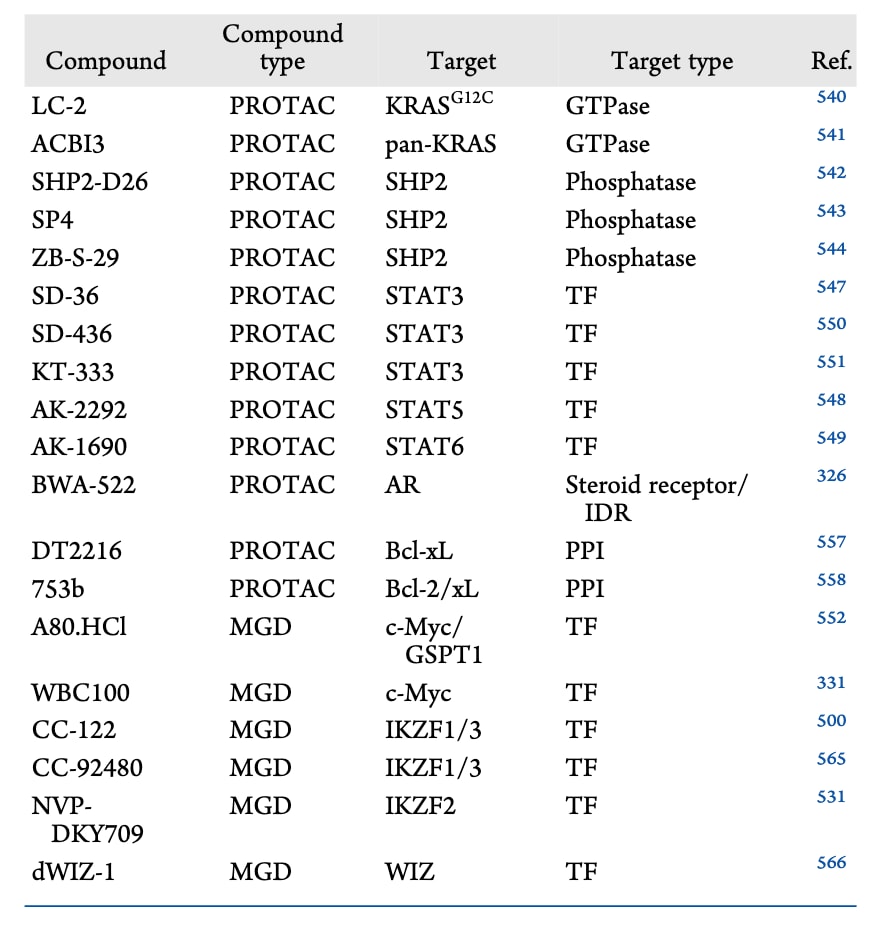
PROTAC通常来源于理性设计,而分子胶降解剂往往最初通过偶然发现获得,因此其理性设计难度更大。不过近年来该领域仍取得了显著进展。例如,通过对CRBN配体进行特定修饰,可以实现对不同底物蛋白的选择性降解。目前已报道多种靶向GSPT1、IKZF1/3、IKZF2、CK1α、ZBTB16、WEE1以及VAV-1的降解剂,其中部分已进入临床研究阶段。
除泛素-蛋白酶体系统外,还存在其他能够实现蛋白降解的策略,用于靶向传统上难以成药的蛋白。例如LYTAC可用于降解膜蛋白和分泌蛋白,而ATTEC和AUTAC则可通过溶酶体途径实现蛋白降解。该综述后续部分主要关注TPD领域的研究进展以及最新的计算辅助TPD技术,并重点讨论其在难成药靶点中的应用。
表5|靶向难成药靶点的代表性降解剂。
5.2 难成药靶点的降解策略
KRAS长期以来被认为是难以成药的靶点,直到Shokat团队发现KRAS
SHP2由于其PTP结构域带有正电荷且在PTP家族中高度保守,一直被视为难成药靶点。SHP2-D26是首个针对SHP2设计的PROTAC,利用VHL配体实现降解,在KYSE520和MV4-11细胞系中的
信号转导与转录激活因子(STAT)家族是参与细胞生长、分化和凋亡等多种生物过程的重要转录因子,也是癌症、炎症及自身免疫疾病的潜在治疗靶点。然而目前尚无直接靶向STAT蛋白的获批药物。Wang团队在STAT降解剂研究方面取得重要进展,例如基于磷酪氨酸模拟抑制剂和CRBN配体设计的STAT3降解剂SD-36,该分子在细胞模型中对STAT3具有高度选择性(相较其他STAT蛋白选择性超过100倍)。随后又开发了STAT5选择性降解剂AK-2292以及STAT6选择性降解剂AK-1690。近期报道的新型STAT3降解剂SD-436在结构上改变了STAT3 warhead的连接位点,将其转移至pro-(R)苯基。临床阶段STAT3降解剂KT-333(NCT05225584)采用与SD-436相同的连接位点,但与SD-36和SD-436不同,KT-333通过招募CRL2
c-Myc是一种在结构上高度无序的转录因子,在多种生物学过程中发挥关键作用,并在多种癌症中过度表达,因此成为重要的抗癌靶点。然而,小分子直接抑制c-Myc一直十分困难,使其长期被认为是难成药靶点。TPD策略的出现为c-Myc干预提供了新的可能。例如A80.2HCl能够同时结合GSPT1和c-Myc,并招募CRBN实现降解,在纳摩尔浓度即可诱导c-Myc降解,同时恢复pRB1蛋白水平并重新建立CDKi耐药细胞系的敏感性。三萜内酯衍生物WBC100则通过E3连接酶CHIP介导c-Myc降解。三萜内酯来源于雷公藤(Tripterygium wilfordii)根部提取的二萜三环氧化合物,具有免疫抑制、抗炎、抗增殖和抗肿瘤等作用,同时也是NF-κB活化的抑制剂。因此WBC100可能通过多种机制发挥抗肿瘤作用。目前WBC100正在中国开展急性髓系白血病治疗的I期临床试验(CTR20243277)。
雄激素受体(androgen receptor,AR)是经过验证的抗肿瘤靶点,目前临床上已使用多种靶向AR配体结合结构域(LBD)的抑制剂。基于AR-LBD结合配体开发的AR PROTAC ARV-766目前正在临床试验阶段(NCT05067140)。然而缺失LBD的构成型活化AR变体会导致对新一代激素疗法(NHA)以及相关PROTAC(如恩杂鲁胺)的耐药。AR的N端结构域(NTD)属于内在无序结构区域,难以被小分子靶向。天然产物EPI-001的发现改变了这一状况,该分子能够与AR-NTD共价结合,从而抑制AR的转录活性并产生抗肿瘤作用。在此基础上进一步开发了针对无序区域配体的PROTAC,例如基于EPI-002设计的BWA-522。该分子采用刚性连接臂并具有良好的口服生物利用度,在实验模型中表现出微摩尔水平的抗增殖活性,为基于IDP配体的PROTAC设计提供了重要起点。
Bcl-2蛋白家族在肿瘤生长和转移过程中发挥重要作用。通过抑制或降解抗凋亡蛋白如Bcl-2、Mcl-1和Bcl-xL,为肿瘤治疗提供了新的策略。然而这些靶点涉及蛋白质−蛋白质相互作用,其界面面积较大,使小分子抑制剂设计面临挑战。Zhou团队开发了首个选择性Bcl-xL降解剂DT2216,该分子基于VHL配体和双重Bcl-2/Bcl-xL抑制剂ABT-263设计。在细胞实验中,DT2216对Bcl-xL的降解
CRBN E3泛素连接酶调节剂(CELMoDs)的一个重要特点是能够降解多种新底物,尤其是含有β发夹环(G-loop)的C2H2锌指转录因子。Lenalidomide通过CRL4
代表性的CELMoD化合物如CC-122和CC92480均以IKZF1和IKZF3为靶点,目前正处于癌症治疗的临床试验阶段(表5)。IKZF1与IKZF2的G-loop仅在一个氨基酸残基上存在差异:IKZF1为Q146,而IKZF2为H141。泊马度胺能够有效降解IKZF1,但不能诱导IKZF2降解,不过仍能促进CRBN与IKZF2的接近。以泊马度胺为起点进一步优化后开发出选择性IKZF2降解剂NVP-DKY709,该分子不影响IKZF1/3,可在体内诱导IKZF2降解并降低Treg抑制功能,目前正在实体瘤的I期临床试验中。
此外,Lenalidomide还能诱导WIZ转录因子的降解。基于这一发现开发的优化降解剂dWIZ-1能够在红系祖细胞中有效诱导胎儿血红蛋白(HbF)表达,为镰状细胞贫血提供潜在的治疗策略。与此同时,通过组合化学构建的CELMoD化合物库进一步扩展了CRBN配体的化学多样性,并扩大了CRBN可降解蛋白质组的范围。
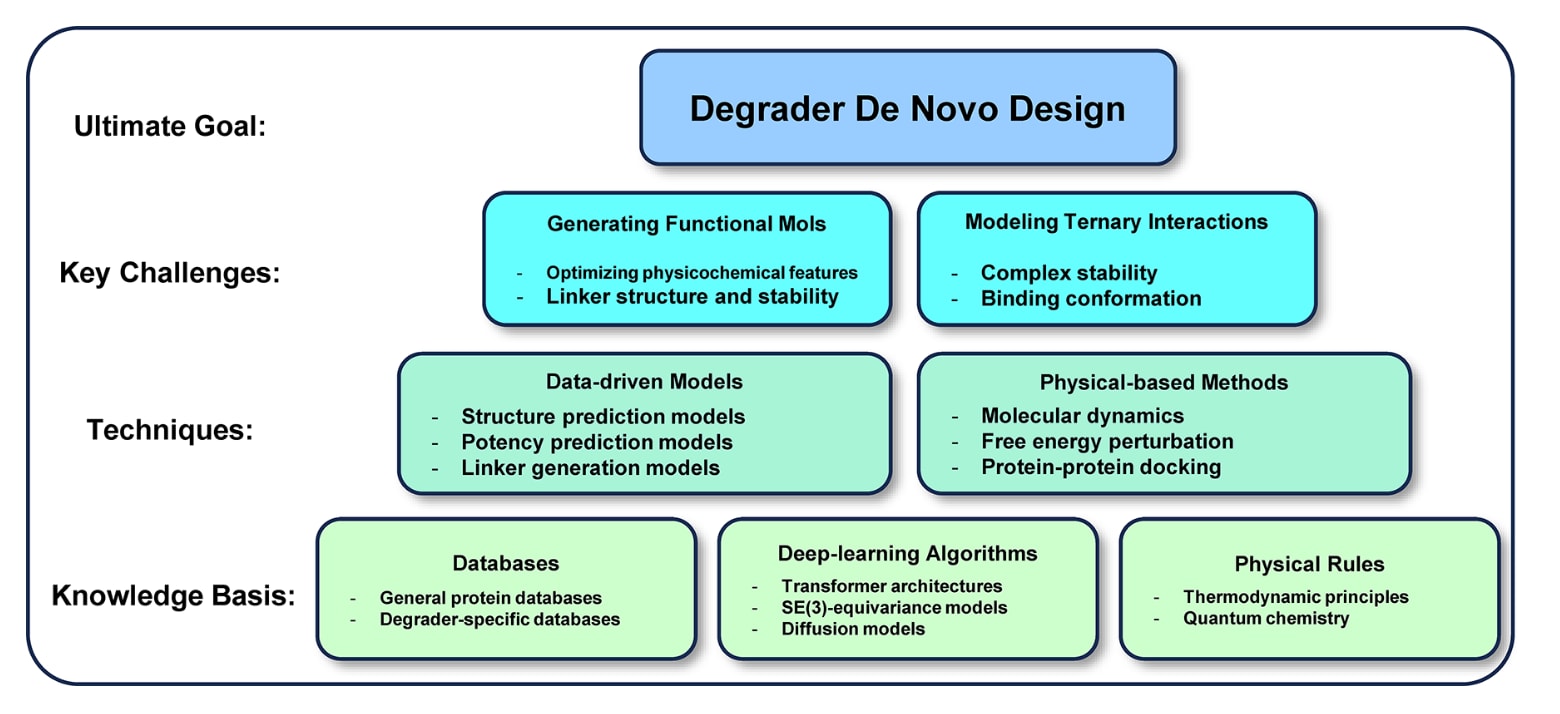
图17|靶向降解剂de novo设计的概念框架。
5.3 计算蛋白降解剂设计
随着靶向蛋白降解领域的持续发展,计算方法已成为加速 PROTAC(蛋白水解靶向嵌合体)及分子胶设计的核心环节。借助物理驱动与数据驱动方法,研究者能够高效探索更广阔的化学空间,筛选出具有潜力的降解剂候选分子,并在实验验证前预测三元复合物的稳定性。下文将围绕数据库构建、连接子设计及相互作用建模三方面,阐述计算技术在其中的作用,及其推动下一代蛋白降解剂设计的潜力与贡献(见图17)。
5.3.1 蛋白降解剂数据库
数据库在发展数据驱动的新型降解剂设计中发挥着关键作用。目前,专门针对 PROTAC 的结构化数据库主要有三个:成熟的 PROTAC-DB 与 PROTACpedia,以及近年建立的 PROTAC-Databank。尽管这些资源在收集和组织 PROTAC 相关数据方面发挥了重要作用,但与通用蛋白质数据库相比,整体规模仍然有限,这对希望充分挖掘 PROTAC 潜力的研究者构成了挑战,尤其在结构与机器学习(ML)应用所需的层面。
PROTAC-DB 是一个持续维护的数据库,整合来自文献的数据并辅以计算特征。在其最新版本(3.0)中,收录了 6111 个 PROTAC 分子、569 种弹头(warhead)、107 种 E3 配体以及 2753 个连接子,并提供详细的生物活性数据,如 DC50、细胞活性,以及渗透性等药代动力学参数。PROTACpedia 则是一个协作型数据库,注册用户可提交结构并访问完整数据集。两者虽有部分条目重叠,但所提供的实验信息可能存在差异,且其数据集规模小于 PROTAC-DB。相比之下,PROTAC-Databank 目前记录的 PROTAC 分子数量较少(3645 个),但其独特之处在于收录了 4142 组靶标-弹头与 E3 连接酶-E3 配体的复合物结构。这种结构导向使 PROTAC-Databank 在计算建模与深度学习应用中尤为宝贵。此外,该数据库基于 DC50 与最大降解速率(Dmax)等指标,为 PROTAC-靶标-E3 连接酶配对引入了降解效率标签,为评估降解性能提供了统一框架。这一标注体系便于其与机器学习流程集成,例如更新的 DeepPROTACs 2.0 模型便利用基于图的架构来预测 PROTAC 降解效率。通过以适配计算工具的格式提供详细结构与生物数据,PROTAC-Databank 有望推动 PROTAC 分子的设计与机制解析。
与 PROTAC 不同,分子胶目前尚缺乏专门的结构化数据库。已报道的分子胶数量相对较少,且作用机制多样,这阻碍了此类资源的建立。部分分子胶诱导的蛋白-蛋白相互作用(MGPPIs)可在通用蛋白-蛋白对接数据集中找到,如 Docking Benchmark 5.5(DB5.5)。这些资源虽可作为评估对接算法的基准,但在为 MGPPIs 建模或训练专用于分子胶的深度学习模型时,仍缺乏所需的特定性与细节。此外,分子胶功效缺乏标准化标签或指标,也限制了这些数据在理性设计中的应用。PROTAC 与分子胶在数据库建设上的差距,凸显了新兴治疗方式在数据可得性方面的普遍挑战。对 PROTAC 而言,将结构、生物与降解数据整合到如 PROTAC-Databank 这样的统一库中,是向前迈出的重要一步,为开发复杂的机器学习驱动设计工具奠定了基础。对分子胶来说,缺乏类似资源则表明,需要系统生成与组织数据,以支持计算与实验发现。随着这些数据库在规模与多样性上不断扩展,它们不仅将提升预测与优化蛋白降解剂特性的能力,还将通过以数据为中心的方法,改变蛋白降解剂设计的整体格局。
5.3.2 PROTAC 连接子设计
PROTAC 连接子的设计面临显著挑战,使其有别于传统小分子药物。不同于常规分子,PROTAC 连接子必须具备可调的柔性、多样的构象可能性以及独特的理化性质,才能发挥功能。即便已知一对合适的配体,要构建出有效的连接子并形成功能性 PROTAC 分子依然困难重重:其一,所设计的连接子需呈现特殊性质,而现有相关数据极为稀少;其二,连接子必须能与配体复杂的三维构象相匹配。
Gharbi 等人总结指出,早期用于 PROTAC 连接子设计的模型包括传统的片段连接方法,如 DeLinker 和 Link-INVENT。这些方法依赖成熟的基于图的算法与生成框架来连接分子片段。然而,PROTAC 连接子因长度更长、配体间初始距离大于典型药物发现中的连接子,带来了独特挑战。即使在这些模型经 PROTAC 专属数据集重新训练后,其表现仍显不足,这凸显了 PROTAC 与常规小分子在设计策略上的根本差异,也表明需要开发专为 PROTAC 连接子定制的模型。
为缓解限制 PROTAC 机器学习模型训练的数据稀缺问题,PROTAC-RL 模型提出新方案:先在一个称为类 PROTAC(quasi-PROTACs)的大型结构数据集上预训练,再在实际 PROTAC 数据上微调。该模型结合增强型 Transformer 架构与强化学习,生成在溶解性、稳定性与生物利用度等关键药代动力学性质上均优化的连接子结构,并已在小鼠实验中得到验证。
下一个挑战是使生成的结构与复杂三维构象对齐。AIMLinker 为此采用门控图神经网络(GGNN),按原子逐步生成连接子,并将输入片段的空间结构信息纳入其中。通过在训练中引入三维特征,AIMLinker 能设计出更符合三元复合物构象需求的连接子。对接与分子动力学模拟验证显示,生成的 PROTAC 具有高结合亲和力与结构完整性。此外,AIMLinker 强大的后处理能力与快速生成速度,提高了构建多样化 PROTAC 候选库的效率。
另一创新模型 PROTAC-INVENT 在 REINVENT 框架基础上,将三维结合位点数据纳入工作流程。与此前主要生成二维结构的模型不同,它整合来自蛋白三元结构(PTS)的三维结合位点数据,以优化生成 PROTAC 在靶标结合口袋中的契合度。其流程分为两步:首先用生成模型产生连接子的 SMILES 字符串,再预测完整 PROTAC 分子的三维结合构象。此优化过程确保与靶标结合位点的更好匹配。
最近,扩散模型的兴起促使其在连接子结构预测中的应用。DiffPROTACs 利用 O(3)-等变图变换器模块,将图神经网络处理空间数据的优势与变换器提取特征的能力相结合,生成 PROTAC 连接子。通过在自建 PROTAC 数据集上微调,该模型实现了 93.86% 的有效率,并为后续研究提供了生成 PROTAC 的综合数据库,缓解了数据稀缺问题,且能生成与 PROTAC 配体构象需求高度契合的连接子。
迄今为止,现有方法仍难以全面兼顾连接子性质、三元复合物稳定性与药代动力学优化之间的相互作用。此外,计算方法常依赖多样性与规模仍有限的数据集,制约了泛化能力。将连接子设计与配体优化相融合,实现两者的协同增强,仍是 PROTAC 从头设计未来一个雄心勃勃但前景可期的发展方向。
5.3.3 分子胶诱导蛋白质−蛋白质相互作用的建模
分子胶诱导的蛋白质−蛋白质相互作用建模仍然是药物发现中一个新兴且复杂的研究方向。与PROTAC三元复合物形成机制已较为清晰不同,目前对分子胶作用机制的理解仍然有限。这种认识不足使得理性设计和数据驱动的开发受到限制,因为分子胶稳定的蛋白质相互作用通常是瞬时的、较弱的并且具有明显的环境依赖性。尽管如此,随着分子对接、分子动力学模拟以及深度学习技术的发展,计算方法已经开始为研究这些相互作用提供重要工具。
蛋白质−蛋白质对接是模拟分子胶诱导PPI的基础方法之一。该方法通过分析蛋白质结构特征,生成候选结合构象(即decoys),以预测两种蛋白之间的相互作用方式。常用工具如HADDOCK和RosettaDock已经被应用于该领域,并能够在对接过程中引入分子胶作为第三方稳定因子。这些工具通过评估在分子胶存在条件下蛋白质表面的互补性,帮助理解弱或瞬时相互作用如何被稳定。然而,仅依赖对接方法难以完全捕捉复合物的动态特征,因为该方法通常基于相对静态的蛋白质结构。
为了进一步优化预测的复合物并分析其稳定性,研究中常在对接之后进行分子动力学(MD)模拟。MD能够反映蛋白质柔性及构象变化,揭示分子胶如何重新组织相互作用界面、识别关键结合热点并评估在生理条件下相互作用的稳定性。此外,自由能计算方法,如自由能微扰(FEP),可以量化分子胶对蛋白质−蛋白质结合热力学性质的影响。尽管这些方法计算成本较高,但能够提供高分辨率的能量学信息,从而帮助筛选优先进行实验验证的候选分子。
深度学习技术的发展也显著提升了分子胶诱导PPI建模能力。早期模型如MaSIF和dMaSIF通过识别蛋白质界面的几何与生化特征,为该领域奠定了基础,而AlphaFold3则通过在统一框架中整合序列、结构和相互作用信息实现了重要突破。AlphaFold3采用基于扩散模型的神经网络架构,可预测复杂生物分子的结构,包括分子胶所涉及的弱或瞬时相互作用。该模型在多种生物分子相互作用预测中表现出前所未有的准确度,即使在存在显著构象变化的情况下也能提供可靠结果。
AlphaFold3能够预测分子胶介导相互作用的重要原因在于其新的训练策略与模型架构。与早期模型不同,AlphaFold3引入pairformer模块用于高效处理分子之间的成对关系,并通过扩散模块描述生物分子的多尺度空间构型。这种结构使模型能够捕捉分子胶诱导的动态结构重排,并揭示目标蛋白与E3泛素连接酶之间的复杂相互作用。实验评估表明,AlphaFold3在界面预测方面的准确度高于传统基于物理模型的方法以及早期机器学习模型。不过仍然存在一些挑战。例如已有数据集主要集中于高亲和力相互作用,这限制了模型对分子胶常见的弱和瞬时PPI的泛化能力。因此,需要构建更符合分子胶特性的专用数据集,以充分发挥AlphaFold3在理性药物设计中的潜力。
5.3.4 降解剂的De Novo设计:潜力与挑战
降解剂(包括PROTAC和分子胶)的de novo设计代表了药物发现中的一种重要发展方向。通过结合计算化学与机器学习方法,可以从传统的试错式合成逐渐转向理性、数据驱动的设计策略,从而优化降解剂的药理特性。然而,要真正实现这一目标仍需解决一系列关键问题。
由于PROTAC相关数据相对丰富,目前已有多种工具可用于其设计。例如DeepPROTACs 2.0虽然并非完全意义上的de novo设计工具,但作为一种高效的降解效率预测模型,能够显著提高降解剂开发效率。该模型能够捕捉蛋白质口袋中的精细相互作用以及分子之间的非共价相互作用。在包含4140个已标注PROTAC的数据集中,其更新后的模型结构达到83.45%的预测准确率和0.9001的AUROC,相比原始模型分别提高约8%和9%。
在新分子生成方面,Nori等人基于GraphINVENT构建了一个从头设计降解剂的生成框架,可同时优化PROTAC的所有分子组成部分。与传统只针对PROTAC某一结构部分进行优化的模型不同,该方法利用强化学习(reinforcement learning,RL)根据预测的降解活性对生成分子进行优化。在以IRAK3为靶点的案例研究中,生成分子的预测活性从50.8%提升至83.8%,显示出较好的设计能力。
近期,Mslati等人开发了用于PROTAC de novo设计的综合计算流程PROTACable。该流程整合了分子对接、机器学习方法(包括SE(3)等变图变换网络)以及实验PROTAC数据库,可生成并评估结构精度较高的三元复合物模型。PROTACable在已解析晶体结构的三元复合物基准测试中表现出优于现有方法的性能。在针对G9A的案例研究中,该系统设计出多种新型VHL型PROTAC,展示了其在药物耐受和难成药靶点研究中的应用潜力。
针对分子胶的de novo设计,Geoffrey等人开发了Molecular Glue-Design-Evaluator(MOLDE)平台。该平台整合蛋白质−蛋白质对接、MD模拟、自由能计算(如MMPBSA和FEP)以及量子力学/分子力学(QM/MM)方法,用于预测和优化分子胶诱导的三元复合物结构。通过对已报道的分子胶体系(例如Protein Data Bank中的相关结构)进行回顾性分析验证,MOLDE能够准确再现结合构象、热力学稳定性以及三元复合物的形成过程。更重要的是,该平台还成功设计出针对DDB1-CDK12体系的新型分子胶,展示出一定的分子生成能力。
尽管这些研究取得了重要进展,但仍然存在多方面挑战。首先是数据数量不足以及数据质量问题,这限制了模型训练和验证的效果。例如PROTAC-DB等现有数据库往往缺乏完整注释,尤其是在三元复合物结构和降解活性指标方面。其次,降解剂功能本身涉及多个相互关联的优化目标,使得建模过程更加复杂。此外,计算预测结果与实验验证之间仍存在一定差距。de novo设计往往产生理论结构,但这些结构可能难以合成或难以在实验条件下实现。与此同时,当前模型多基于静态结构表示,难以充分考虑降解过程中关键的动态构象变化。
未来研究需要进一步扩展数据集规模并提高数据质量,同时引入真实时间尺度的动力学信息以提高模型准确度。多目标优化算法的发展、实验反馈的持续整合以及三维结构感知建模与迁移学习方法的应用,将有助于探索新的降解剂化学空间,从而推动难成药靶点治疗策略的发展。
6 结论与展望
“不可成药”靶点通常具有独特特征,例如缺乏配体结合口袋、高度保守的活性位点、功能由蛋白质-蛋白质相互作用(PPI)介导,以及缺乏稳定或明确的三维结构。药物发现与开发技术的进步,以及基于物理和人工智能的计算方法,催生了应对这些高难度靶点的创新策略。针对固有无序蛋白(IDP)与 PPI 的干预方法、变构药物发现以及靶向蛋白降解等领域已显现出重大潜力。这些进展促成了多个先导化合物的诞生,部分甚至已获临床批准。因此,“不可成药”的概念正逐渐转变为“难以成药”或“尚未成药”,反映出应对这些一度棘手的靶点的能力正在不断增强。
该综述重点介绍了针对不可成药蛋白的小分子药物发现策略,特别强调了计算方法在其中发挥的重要作用,并总结了已开发的成功及潜在治疗实体。尽管创新药物发现技术与新型治疗实体的出现带来了令人振奋的可能性,但知识空白依然显著,持续研究以克服这些挑战十分必要。
IDP 在人类蛋白质组中广泛存在,且在多种疾病的发病机制中扮演关键角色。将 IDP 转化为可成药靶点,将大幅拓展可成药靶标空间。虽然针对 IDP 的药物设计方法已经问世,但仍面临重大挑战与局限。成药 IDP 的主要障碍在于缺乏优化 IDP 配体的高效方法,这需要具备更高时空分辨率的实验技术,以及能有效分析 IDP-配体动态结合模式的新颖计算手段。以下方向有望加快 IDP 药物发现:(1)AI 辅助药物设计。鉴于 IDP 配体具有“多构象亲和性”特征,多靶点药物设计 AI 模型在生成新型 IDP 配体方面潜力巨大。(2)配体特异性预测与验证。IDP 配体结合模式复杂,且受 IDP 靶标调控的互作网络广泛,其特异性与调控精确性备受关注。实验上建议采用如 MS-CETSA 等蛋白质组学技术评估脱靶效应;计算上,基于 AI 的多组学模型有望预测 IDP 配体对细胞内分子网络的影响,但仍需深入探索。(3)凝聚体调节疗法(C-mods)。靶向无膜细胞器或生物分子凝聚体,是应对不可成药蛋白的有力策略。许多 IDP(包括关键转录因子)通过凝聚体形成推动疾病进展。已有报道指出,小分子配体可通过诱导形成、促进溶解、改变定位或调节理化性质等方式调控疾病相关凝聚体。通过作用于功能过程而非直接结合或降解蛋白,C-mods 有望提供更有效且更具选择性的治疗方案,并减少副作用。
变构药物发现在攻克不可成药靶点方面已取得显著成功。变构位点预测与变构药物设计的进步促成了大量变构调节剂的发现,其中部分已上市。然而,仍需应对若干挑战以进一步加速该领域发展。例如,(1)变构调控机制有待深入探索。一旦发现变构位点,预测结合化合物的调控强度上限及其作为激活剂、抑制剂或中性占位者的作用属性仍具难度。变构理论、计算方法、扩展的实验数据集以及 AI 驱动模型的进展或可提供帮助。(2)亟需开发高效的隐藏变构位点发现方法。隐藏变构位点常通过高通量筛选或长期分子动力学模拟偶然发现,耗时较长。现有计算方法大多仅能预测单体蛋白中的隐藏位点,且在非结合态结构中的预测准确性偏低。迫切需要更快、更准确且更全面的隐藏变构位点预测方法。此外,探究隐藏变构位点形成的进化机制亦值得深入研究。
随着新药物靶点的不断涌现与结构研究的进展,潜在治疗靶标空间持续扩大,甚至延伸至非蛋白类靶点。据估计,人类基因组约 75% 可转录为 RNA,而仅有 1.5% 编码蛋白质。RNA 靶向,尤其是小分子干预 RNA,是药物发现中相对新颖且快速发展的领域。潜在 RNA 靶点包括编码被归为不可成药的疾病相关蛋白的 mRNA,以及影响疾病进程的非编码 RNA。尽管功能重要,RNA 作为药物靶点的潜力仍未充分发掘。这主要源于其固有挑战:内在柔性、强负电荷以及缺乏明确的疏水口袋。RNA 与小分子的相互作用主要由非共价力主导,如堆叠作用和氢键。芳香族小分子可嵌入碱基对之间增强 π 相互作用,而 RNA 带负电的糖-磷酸骨架有利于与带正电配体的静电作用。因此,现有化合物库筛选常得到性质相似、非特异性结合且具有毒性的分子。应对之道在于开发针对 RNA 靶点的专用化合物库与定制筛选方法。技术进步已催生特定策略,如核糖核酸酶靶向嵌合体(RIBOTAC),可招募 RNase-L 促进 RNA 的靶向降解。高通量筛选常获得因与非功能区结合而生物学惰性的 RNA 结合化合物,幸运的是,RIBOTAC 等方法可将此类配体转化为有效工具。
从可成药化学空间的角度看,开发用于高通量筛选的专用化合物库与应用 AI 辅助从头药物设计至关重要。如前所述,RNA 靶向筛选的一大挑战是频繁识别化学骨架相似的基团,导致特异性低。由于多数小分子库面向蛋白靶点设计,亟需构建 RNA 偏好配体库。此外,PPI 调节剂与 PROTAC 往往不符合 Lipinski 五规则等传统指导原则。现有商业化合物库多为传统靶点设计,未能覆盖不可成药靶点的配体化学空间。因此,必须通过新技术拓展化学空间至未探索领域。一方面,自动化实验技术的迅速普及促进了高通量合成构建化合物库的方法。然而,在打造化学多样性丰富的类药分子库,尤其是面向特定靶点的聚焦库并与机器学习结合实现快速活性筛选方面,仍存在重大挑战。另一方面,基于 AI 的深度生成模型为从头药物设计带来新机遇。依赖已知活性化合物预测新分子活性的深度生成模型局限于狭窄的化学空间,而以靶标结构为导向的从头生成模型则可探索更广的化学空间。为确保生成分子可被实验测试,扩展的化学空间必须保持合成可及性。这要求进一步整合分子生成与合成路线规划,以及反应产率与条件的预测建模。
尽管该综述聚焦于针对不可成药靶点的小分子药物设计,AI 辅助蛋白质结合剂设计这一新兴领域值得更多关注。随着 RFdiffusion、Chroma、SCUBA-D 等新蛋白质支架生成工具,以及 ProteinMPNN、ABACUS-R、ProDesignLE、GeoSeqBuilder 等序列生成方法的出现,能够结合 PPI、IDP 乃至 RNA 的蛋白质可被从头设计。已有报道指出具备治疗潜力的蛋白质结合剂,其中部分甚至可口服给药。同时,肽类尤其是环肽也为不可成药靶点提供了新机遇。尽管蛋白质结合剂易于作用于细胞因子、膜锚定受体等分泌蛋白,其细胞内递送仍需深入研究。还应注意到,通过表型筛选与 AI 建模绕过靶点特异性难题的可能性。例如,化学扰动基因表达谱与单细胞 RNA 测序数据已被用于在不明确细胞靶标的情况下预测新化合物的效应。深度学习模型仅凭蛋白质序列与配体化学结构即可预测蛋白-配体相互作用,无需显式的蛋白质结构数据。实验筛选与主动学习框架的结合已促成新型抗生素及抗衰老化合物的发现。
随着对疾病机制理解的加深,潜在药物靶标空间预计将进一步拓展。为应对新药靶点与既有难点靶标的成药挑战,持续开发创新型计算机辅助药物设计方法,并与新兴药物发现及开发技术并行推进,至关重要。凭借这些进展,大多数不可成药靶点终将有望进入可治疗干预的范畴。