Nat. Commun. 2026 | EPMolGen: 深度学习辅助发现一种高效且具有细胞活性的RNA N6-甲基腺苷识别蛋白YTHDC2抑制剂
今天介绍的是发表在 Nature Communications 上的一项研究工作。RNA中N6-甲基腺苷(m6A)修饰在多种生理与疾病过程中发挥关键作用,而其识别蛋白YTHDC2被认为是重要的潜在治疗靶点,但一直缺乏高效的小分子抑制剂。针对这一问题,研究提出了EPMolGen模型,在分子生成过程中显式引入蛋白质静电特征,从而提升生成分子的结合能力与药物性质。实验结果显示,该模型在QED、合成可及性等关键指标上优于现有方法,并成功筛选出活性化合物H3。进一步通过结构优化获得高效抑制剂DC2-C1,其IC50达到0.168 μM,并在细胞实验中表现出良好的靶向活性与选择性。该研究展示了深度学习在药物早期发现中的应用潜力,为靶向YTHDC2的药物开发提供了重要线索。

获取详情及资源:
0 摘要
YTHDC2是一种独特的含YTH结构域蛋白,能够识别RNA上的N6-甲基腺苷(m6A),在多种病理过程中发挥关键作用,并被视为具有潜力的治疗靶点。然而,迄今尚未有高效的小分子抑制剂被报道。
为弥补这一空白,构建了一种基于深度学习的分子生成模型EPMolGen,该模型显式引入受体蛋白的静电特征。在计算验证中,该模型达到了当前最先进的性能水平。利用EPMolGen,筛选得到YTHDC2抑制剂H3,其IC50为16.84 μM。在此基础上对H3进行结构优化,获得了高效化合物DC2-C1,其针对YTHDC2的IC50为0.168 μM,并对其他YTH结构域蛋白表现出良好的选择性。
在细胞实验中,DC2-C1能够有效作用于YTHDC2。进一步研究表明,DC2-C1处理可显著降低多个YTHDC2靶mRNA的表达水平,从而抑制相关细胞的表型表现。
整体而言,该研究展示了深度学习在药物发现中的巨大潜力,并为靶向YTHDC2的药物开发提供了具有前景的先导化合物。
1 引言
N6-甲基腺苷(m6A)是真核生物中常见的一种RNA修饰。其通过甲基转移酶(“写入器”)、去甲基化酶(“擦除器”)以及m6A识别蛋白(“读取器”)的协同作用,动态调控RNA的命运与功能。这种调控机制影响多种生物学过程,包括细胞分化、发育及疾病进展。
在上述组分中,m6A读取器作为效应蛋白,参与多种生理与病理过程。目前,含有YT521-B同源结构域(YTH)的蛋白(包括YTHDC1-2与YTHDF1-3)被认为是m6A修饰mRNA的主要读取器。
YTHDC2是一种独特的含YTH结构域的m6A读取蛋白,分布于细胞核与细胞质中。其包含多个功能结构域,其中YTH结构域负责识别并结合m6A修饰的RNA分子。YTHDC2在RNA代谢、细胞功能及发育等多种生物过程中发挥关键作用,并与多种疾病的发生发展密切相关。例如,多项研究表明,YTHDC2与类风湿性关节炎的发生与进展密切相关。在该疾病中,类风湿性关节炎成纤维样滑膜细胞是滑膜组织异常增生与侵袭的重要组成部分,其异常增殖与侵袭性表型会加剧关节破坏。抑制YTHDC2在这些细胞中的m6A结合功能,可降低其靶基因AMIGO2的mRNA稳定性,从而减少AMIGO2蛋白表达,进一步抑制细胞的增殖、迁移与侵袭,进而缓解疾病症状。
此外,YTHDC2在肿瘤中的作用具有复杂性,可通过多种机制发挥促癌或抑癌作用。例如,在肝细胞癌与乳头状甲状腺癌中表现为抑癌因子,而在胃癌、胰腺导管腺癌、前列腺癌、鼻咽癌及皮肤鳞状细胞癌中则表现为致癌因子。同时,YTHDC2还参与代谢调控过程,如肝脏脂质生成,从而与部分代谢性疾病的发生相关。
当前对YTHDC2生物学功能的研究主要依赖于基因敲低、敲除或过表达等手段,但这些方法无法区分观察到的效应是否源于其m6A识别功能的阻断。针对YTH结构域的小分子抑制剂有望成为解决这一问题的理想工具。目前仅有一种广谱YTH蛋白抑制剂被报道,其对YTHDC2的抑制活性较弱,IC50为30 μM。因此,开发高效且具有选择性的YTHDC2抑制剂具有重要意义。
传统上,针对新靶点蛋白筛选活性化合物通常依赖高通量筛选方法,该方法成本高且耗时较长。近年来,基于深度学习的生成模型在语音处理、计算机视觉及自然语言处理等领域取得显著进展,并逐步应用于药物发现领域,催生了一系列用于分子生成的专用模型。其中,基于受体结构的分子生成模型(如LiGAN、Pocket2Mol、GraphBP、TargetDiff与PocketFlow)代表了当前前沿方法。
LiGAN通过将蛋白-配体复合物转化为三维原子密度网格,并利用三维卷积神经网络进行分子生成。Pocket2Mol采用E(3)等变生成框架,在捕捉分子图拓扑与几何约束方面优于非等变模型。GraphBP引入局部球坐标系以参数化空间中的分子生成过程。TargetDiff构建了面向靶点的全原子三维等变扩散模型。PocketFlow则基于自回归流模型,并引入化学知识,其生成分子的生物活性已在实验中得到验证。
尽管上述模型在基准测试中表现优异,但仍存在局限。例如,LiGAN缺乏等变性,其基于三维卷积网络处理离散化原子密度网格的方法限制了模型性能;此外,将连续三维空间离散化为网格的过程也降低了实际应用能力。LiGAN、GraphBP与TargetDiff依赖Open Babel进行化学键生成,这种后处理方式可能产生不理想的结构,降低分子的类药性并增加合成难度。除PocketFlow外,大多数模型仅在计算环境中验证,缺乏实验验证这一关键环节。此外,这些模型均未显式考虑靶蛋白的静电特征(如电场强度与电势),而这些因素在配体-蛋白相互作用中至关重要。
针对上述问题,构建了EPMolGen模型,这是一种基于自回归流的深度生成模型,在架构中显式引入靶蛋白的静电特征。通过一系列计算实验验证了其有效性。进一步利用该模型生成针对YTHDC2的分子,并通过实验筛选获得活性化合物H3。在此基础上进行结构优化,最终获得具有高活性且具备细胞作用能力的候选化合物。
2 结果
2.1 EPMolGen生成模型的构建
EPMolGen是一种基于自回归流的深度学习模型,专为结合口袋感知的分子生成任务设计。与传统分子生成方法不同,该模型显式引入受体蛋白的静电特征,而这些特征是配体-受体相互作用的关键决定因素。通过嵌入这些信息,模型在生成具有生物活性的分子方面的能力得到增强。
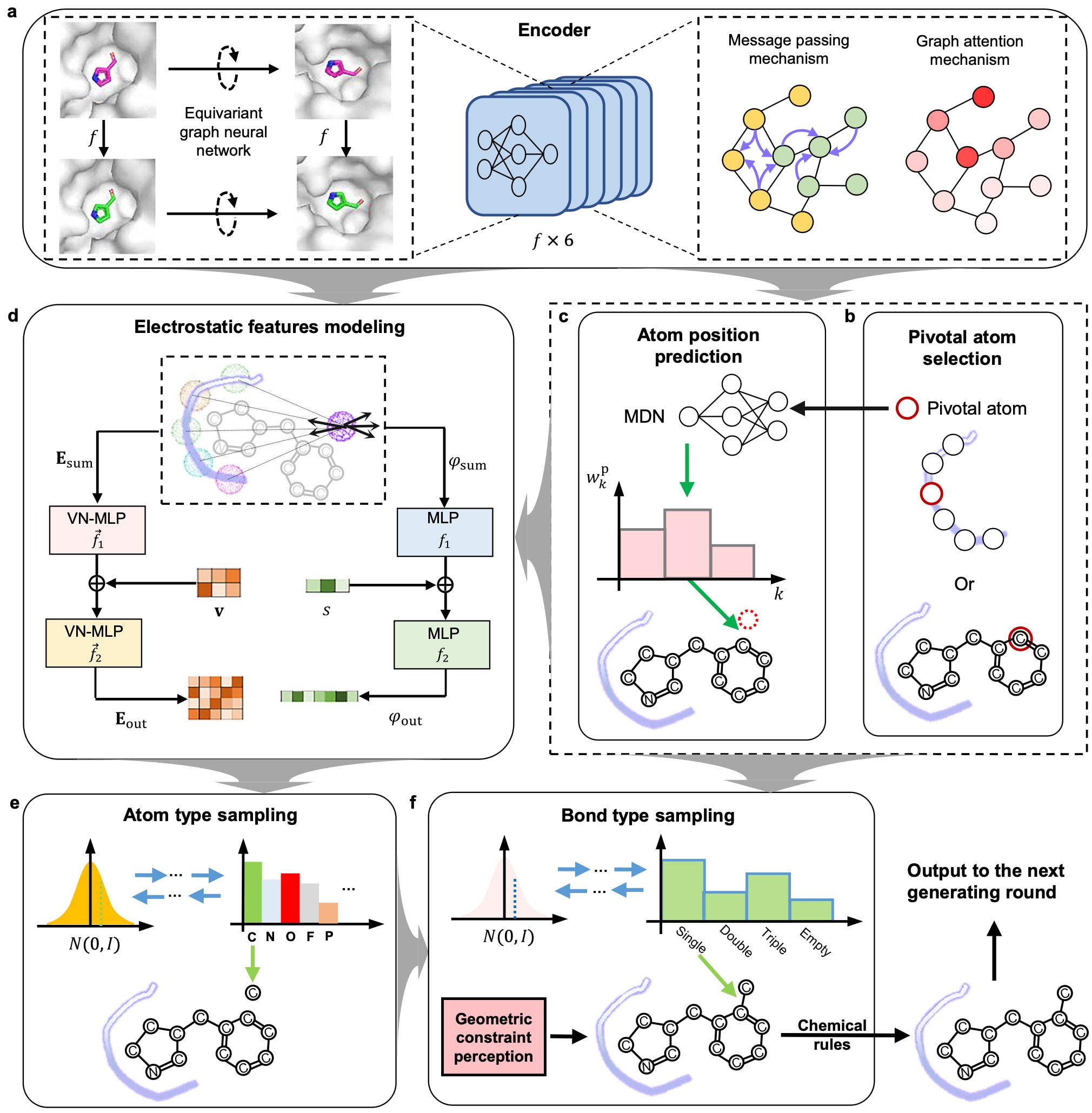
图1|EPMolGen 的结构示意图 a. 编码器模块,其中 f 表示等变图神经网络。黄色与浅绿色圆分别表示 KNN(k近邻)图中的蛋白原子节点与配体原子节点。红色深浅变化表示不同的注意力权重。蛋白质与分子结构由 PyMOL(Version: 3.1)绘制。 b. 关键原子选择模块,紫色曲线表示蛋白口袋。 c. 原子位置预测模块,其中 k 表示不同的高斯混合分量,wkp 表示对应分量的系数。 d. 静电特征建模模块,Esum 和 φsum 分别表示在预测原子位置处由蛋白口袋所有原子贡献的总电场强度与电势。v 和 s 分别表示新预测原子位置的向量与标量上下文特征,来源于编码器模块输出。 e. 原子类型采样模块,用于采样下一个生成原子的类型,基于自回归流,仅包含9种可能原子类型(C、N、O、F、P、S、Cl、Br 和 I)。 f. 键类型采样模块,用于采样化学键类型,同样基于自回归流,仅包含4种可能键类型(单键、双键、三键和空键),其中空键表示两个原子之间不存在键。
在具体实现中,采用点电荷模型对蛋白质进行建模,原子电荷来源于OPLS4力场。在结合口袋中任意位置的电场强度与电势,通过对所有点电荷的贡献进行求和得到,从而实现对蛋白质静电环境的物理准确描述。
EPMolGen的整体流程由六个模块构成。首先为编码模块,该模块利用三维等变神经网络对蛋白口袋或蛋白-配体复合物进行表征,并结合消息传递与图注意力机制,捕捉结合位点的空间结构与环境信息。其次为关键原子选择模块,根据编码结果选择一个关键原子;在空口袋情况下,从蛋白原子中选择,在蛋白-配体复合物中则从已有配体原子中选择。
第三为原子位置预测模块,采用混合密度网络,根据编码信息与关键原子特征预测新原子的位置。该模块仅考虑距离关键原子2 Å范围内的位置。第四为静电特征建模模块,用于提取电场强度与电势表示,并与环境特征进行融合。
最后两个模块分别负责原子类型采样与化学键采样。在前述模块生成的特征引导下,完成新原子与化学键的生成。其中,化学键类型采样模块引入几何约束与化学规则,以确保生成结构的合理性。为建模几何约束,采用受AlphaFold2启发的三角自注意力机制,用于刻画分子图中的三边关系。例如,当原子i与j之间存在三键时,另一原子k同时与二者形成双键的概率将显著降低。此外,仅允许与新生成原子距离4 Å以内的已有配体原子形成化学键。
在化学合理性方面,模型会剔除违反原子价规则或形成不合理结构(如O-O、O–N、C = C = C及三元环)的候选键,并重新采样直至满足约束。通过这一机制,确保生成分子符合基本化学原理。最终,新生成原子的空间位置、类型及其化学键被整合进当前分子环境,作为下一步生成过程的输入。
2.2 EPMolGen性能评估
通过选取10个具有代表性的靶蛋白测试集对EPMolGen性能进行评估,这些靶点曾用于评估其他生成模型,其PDB编号分别为1bvr、1u0f、1zyu、2ah9、2ati、2hw1、4bnw、4i91、5g3n和5lvq。针对每个靶蛋白的结合口袋,利用EPMolGen生成10,000个分子,并计算其多项关键性质的平均值,包括化学结构有效性、类药性定量评分(QED)、辛醇-水分配系数(logP)以及合成可及性(SA),上述指标均通过RDKit工具计算获得。
为对EPMolGen进行基准测试,同时评估了多种代表性生成模型,包括LiGAN、Pocket2Mol、GraphBP、TargetDiff和PocketFlow。此外,还计算了CrossDocked2020数据集中分子的对应性质作为参考基线。
表1|不同生成模型生成分子及 CrossDocked2020 数据集中分子四项关键性质的平均值计算结果
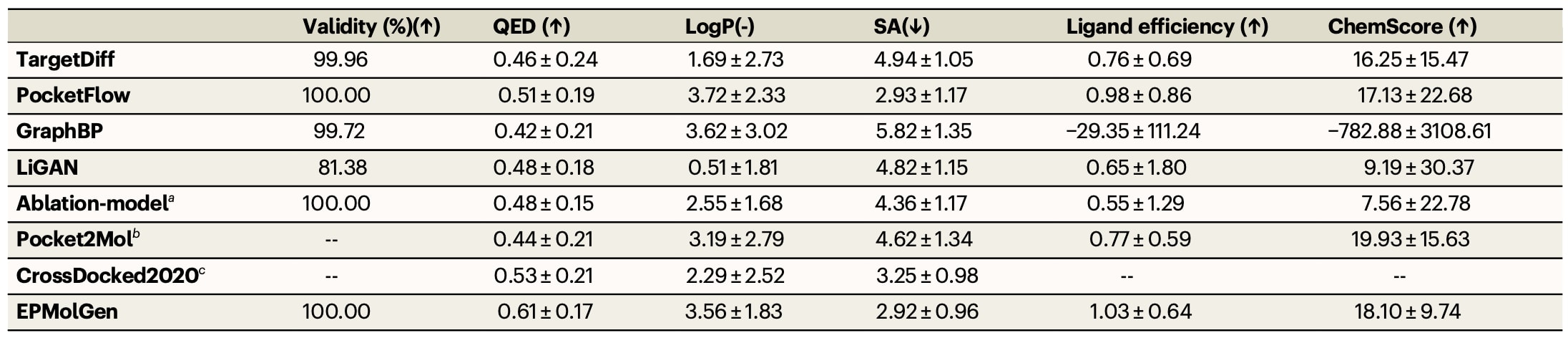
a. 消融模型与 EPMolGen 结构相似,但未显式考虑蛋白靶标的静电特征。 b. Pocket2Mol 在部分靶蛋白上未能生成预期数量的分子,统计分析仅基于成功生成的分子进行。 c. 在计算过程中,已去除 CrossDocked2020 数据集中重复的配体。数据以均值 ± 标准差表示。
不同模型生成分子的关键性质平均值结果如表1所示。计算过程中,首先对每个模型生成的所有分子逐一计算各项指标,然后统计其均值与标准差。结果显示,EPMolGen与PocketFlow生成的分子均达到100%化学有效性,而TargetDiff、GraphBP与LiGAN未能实现完全有效。在logP指标方面,所有模型生成分子的平均值均落在理想范围0–5之间,但在具体靶点上,部分基线模型未能稳定维持在该范围内。
在QED与SA两个关键指标上,EPMolGen表现出显著优势。QED用于衡量分子的类药性,其值越高表示性质越优;EPMolGen生成分子的平均QED显著高于其他模型。SA用于评估分子的合成难度,其值越低表示更易合成;EPMolGen生成分子的平均SA值最低。此外,从分布分析结果可见,该模型在QED与SA上的标准差较小,说明其性能更加稳定一致。
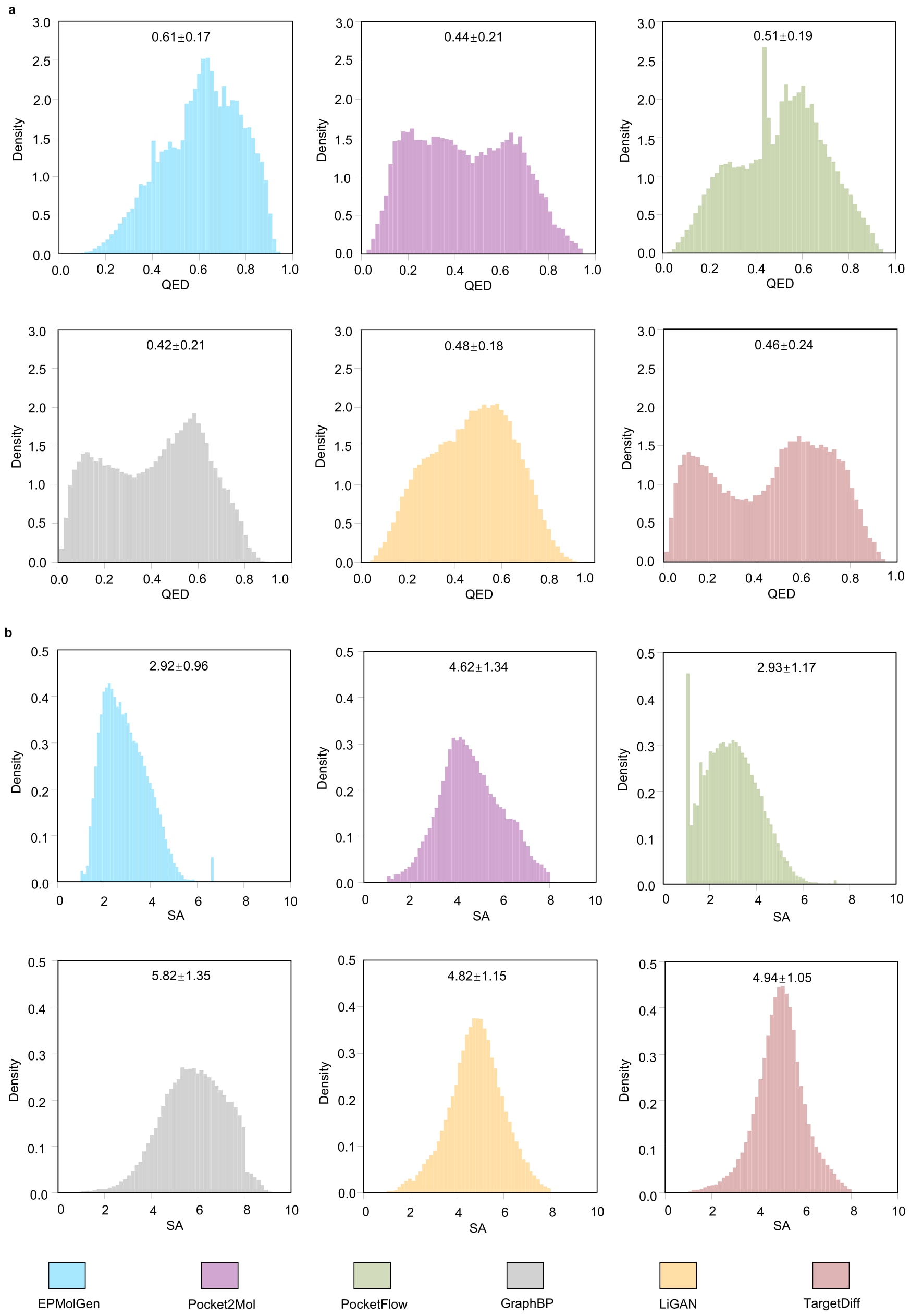
图2|QED 与 SA 的密度分布 a. EPMolGen、Pocket2Mol、PocketFlow、GraphBP、LiGAN 和 TargetDiff 生成分子的 QED 密度分布。 b. EPMolGen、Pocket2Mol、PocketFlow、GraphBP、LiGAN 和 TargetDiff 生成分子的 SA 密度分布。数据以均值 ± 标准差表示。
进一步通过ChemScore评估生成分子的结合亲和力。结果表明,EPMolGen、PocketFlow、TargetDiff和Pocket2Mol均表现出较高的平均结合亲和力。GraphBP则出现异常值,可能由于其生成分子与蛋白发生空间冲突或未位于结合口袋内。考虑到分子量对结合亲和力的影响,引入配体效率(LE)作为补充指标,用于衡量单位重原子的结合能力。结果显示,EPMolGen在所有模型中具有最高的平均LE值,表明其在结合效率方面表现最优。
最后,对生成分子中不常见子结构的比例进行分析,包括三元环、八元环、异常环结构以及由四个及以上稠合环构成的结构。这类结构虽然在部分药物中存在,但通常伴随较高毒性或合成难度。结果显示,EPMolGen在三元环、八元环及异常环结构的生成比例方面均为最低,其整体水平接近参考数据集CrossDocked2020。相比之下,其他模型在不常见结构生成方面表现较差,例如Pocket2Mol生成的分子中有52.6%包含稠合环结构,GraphBP生成分子中84.9%包含三元环且64.2%包含异常环,LiGAN对应比例分别为50.9%与32.2%,TargetDiff则有7.7%的分子包含八元环结构。
表2|不同生成模型生成分子及 CrossDocked2020 数据集中分子中三元环、八元环、稠合环(≥4环融合)及异常环的占比
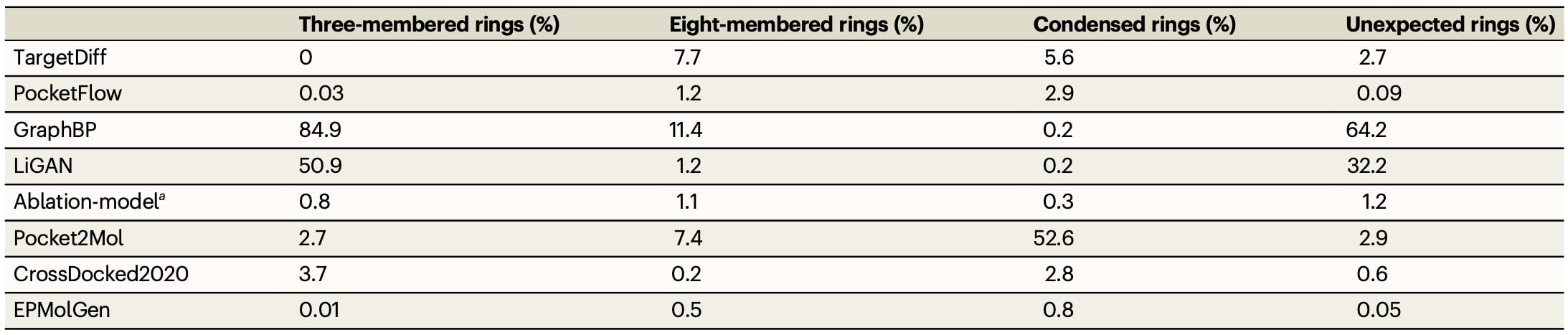
a. 消融模型与 EPMolGen 结构相似,但未显式考虑蛋白靶标的静电特征。 b. 在计算过程中,已去除 CrossDocked2020 数据集中重复的配体。
2.3 消融分析
与已有分子生成模型相比,该模型的一个关键区别在于显式引入了静电特征。为评估该特征对模型性能的影响,进行了消融实验。具体而言,构建了一个不包含静电特征的对照模型(称为ablation-model),并采用与前述相同的方法,对其生成分子的各项性质进行计算与评估。
结果表明,该对照模型同样能够生成化学结构有效且具有合理logP值的分子,同时生成包含不常见子结构的比例较低。然而,在多个关键指标上,其性能明显劣于EPMolGen,包括平均QED、SA、配体效率(LE)以及ChemScore值。尤其是在LE与ChemScore方面,对照模型的表现显著下降。
进一步通过QED、SA、LE及ChemScore的分布分析可见,对照模型整体性能显著低于EPMolGen。综合来看,消融实验结果表明,引入静电特征能够有效提升模型性能,尤其在增强分子与靶点结合能力方面具有重要作用。
2.4 EPMolGen在YTHDC2靶点上的应用获得活性化合物
进一步利用EPMolGen开展针对m6A读取蛋白YTHDC2中YTH结构域的小分子抑制剂筛选。受体三维结构来源于YTHDC2的YTH结构域晶体结构(PDB ID: 6K6U),并以其m6A-mRNA底物的结合位点作为分子生成的目标口袋。基于该模型共生成5,000个候选分子,从中筛选QED值大于0.8的分子,并按照配体效率(LE)排序。在排名前十五的分子中,进一步选取五个(H1–H5)具有较好合成可行性的候选分子进行合成与实验验证。
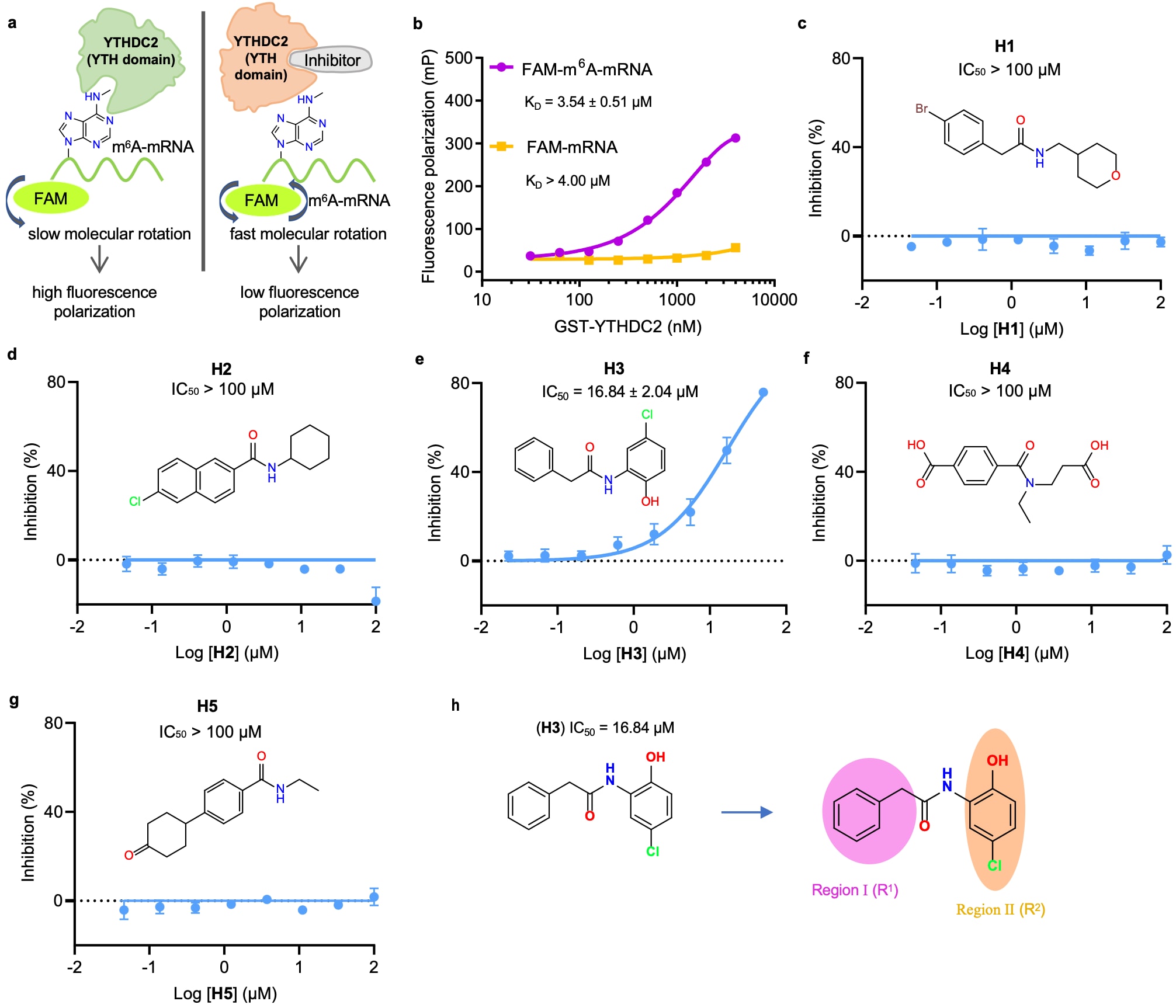
图3|YTHDC2 命中化合物的发现 a. YTHDC2 小分子抑制剂筛选方法(FP)的示意图。 b. 使用 FAM 标记的 mRNA/m6A-mRNA 与 GST-YTHDC2 孵育的 FP 实验结果。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 c–g. 化合物(H1–H5)针对 YTHDC2 的剂量-活性曲线,基于 FP 实验测定。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 h. 用于 H3 结构优化的区域。
为评估这些化合物对YTHDC2的生物活性,表达并纯化了重组人源YTHDC2 YTH结构域蛋白。基于该结构域对m6A的特异性识别能力,建立了以荧光素标记(FAM)的m6A-mRNA为探针的荧光偏振检测体系。在实验过程中,选择使用带GST标签的YTHDC2-YTH蛋白而非去标签蛋白,原因在于后者分子量较小(小于16 kDa),信号强度不足。实验结果表明,GST标签蛋白不仅显著提高了荧光偏振信号,而且不影响YTH结构域对m6A的特异性识别。
在EPMolGen生成的五个候选化合物中,仅H3表现出明显抑制活性,其IC50为16.84 ± 2.04 μM。随后,以H3为基础开展了结构优化及构效关系分析。
2.5 H3的结构优化与构效关系分析
对H3的结构优化主要集中在两个区域:苄基部分(R1)与4-氯苯酚部分(R2)。首先在保持R2不变的情况下,对R1进行优化,共合成了13个在R1位点引入不同取代基的新化合物(14a–m)。其生物活性结果表明,在苯环不同位置引入氟取代(14a–c)未能提升活性,但对位氟取代相对更可耐受。随后将对位氟替换为氯或溴(14d–e),仍未观察到活性提升。在对位引入叔丁基或苯基(14f–g)则导致活性完全丧失。相比之下,引入酯基或羧基(14h–i)能够维持甚至提升活性。
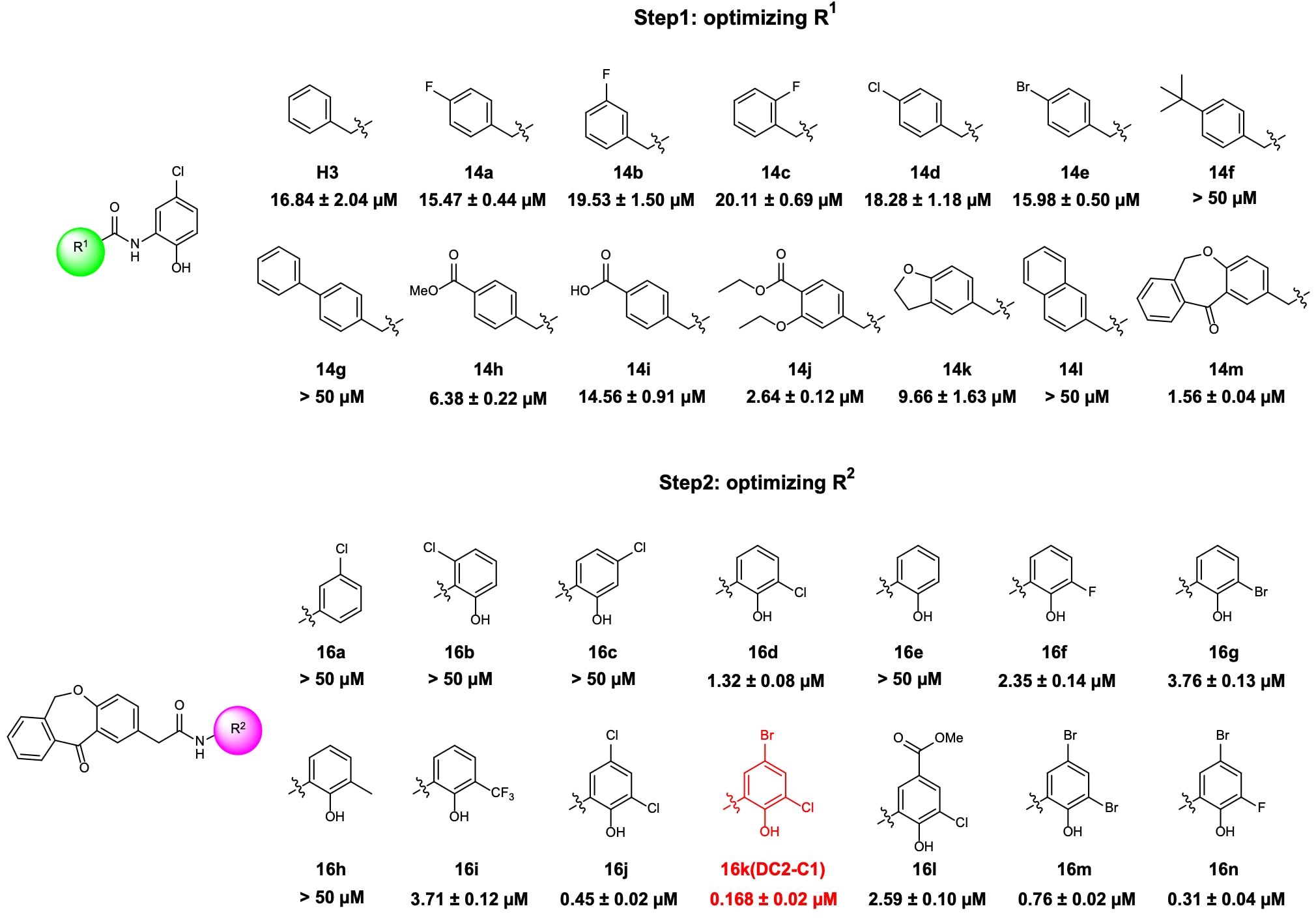
图4|化合物14a–m与16a–n的化学结构及其在FP实验中的生物活性 数据以三次独立实验的均值 ± 标准差表示。
进一步在苯环上引入双取代(包含酯基)后,化合物14j的活性较原始命中化合物H3提高约6倍。具有对位和间位取代的环化产物14k同样表现出优于H3的活性。然而,引入萘环结构的化合物14l未显示活性。值得注意的是,当R1被二苯并[b,e]氧杂卓类似结构(14m)取代时,活性进一步显著提升,其IC50达到1.56 μM,表明该结构是R1位点的优选取代基。
在第二步优化中,固定R1为二苯并[b,e]氧杂卓结构,对R2中的苯基部分进行修饰,共合成14个新化合物(16a–n)。活性测试结果表明,去除苯酚羟基(16a)会导致活性完全丧失,说明该基团对活性至关重要。随后对氯取代位置进行调整或移除(16b–e),发现邻位氯取代(16d)可略微提升活性,而其他变化则导致活性下降或消失。将氯替换为氟(16f)、溴(16g)、甲基(16h)或三氟甲基(16i)均降低了活性。
值得注意的是,在苯环4位和6位同时引入氯原子(16j)可使活性提升约3倍。在此基础上,进一步在该区域引入不同取代基,合成了4个新化合物(16k–n)。其中,R2位点为4-溴-6-氯苯酚结构的化合物16k(命名为DC2-C1)表现出最高活性,其IC50为0.168 μM。
2.6 DC2-C1针对YTHDC2的生物活性表征
通过对初始命中化合物H3进行结构优化与构效关系分析,获得了优化产物DC2-C1。该化合物在荧光偏振实验中表现出显著的抑制活性,其IC50为0.168 ± 0.02 μM,相较于初始化合物H3实现了约100倍的活性提升。在相关实验中,荧光信号强度保持稳定,未观察到化合物引起的荧光干扰。
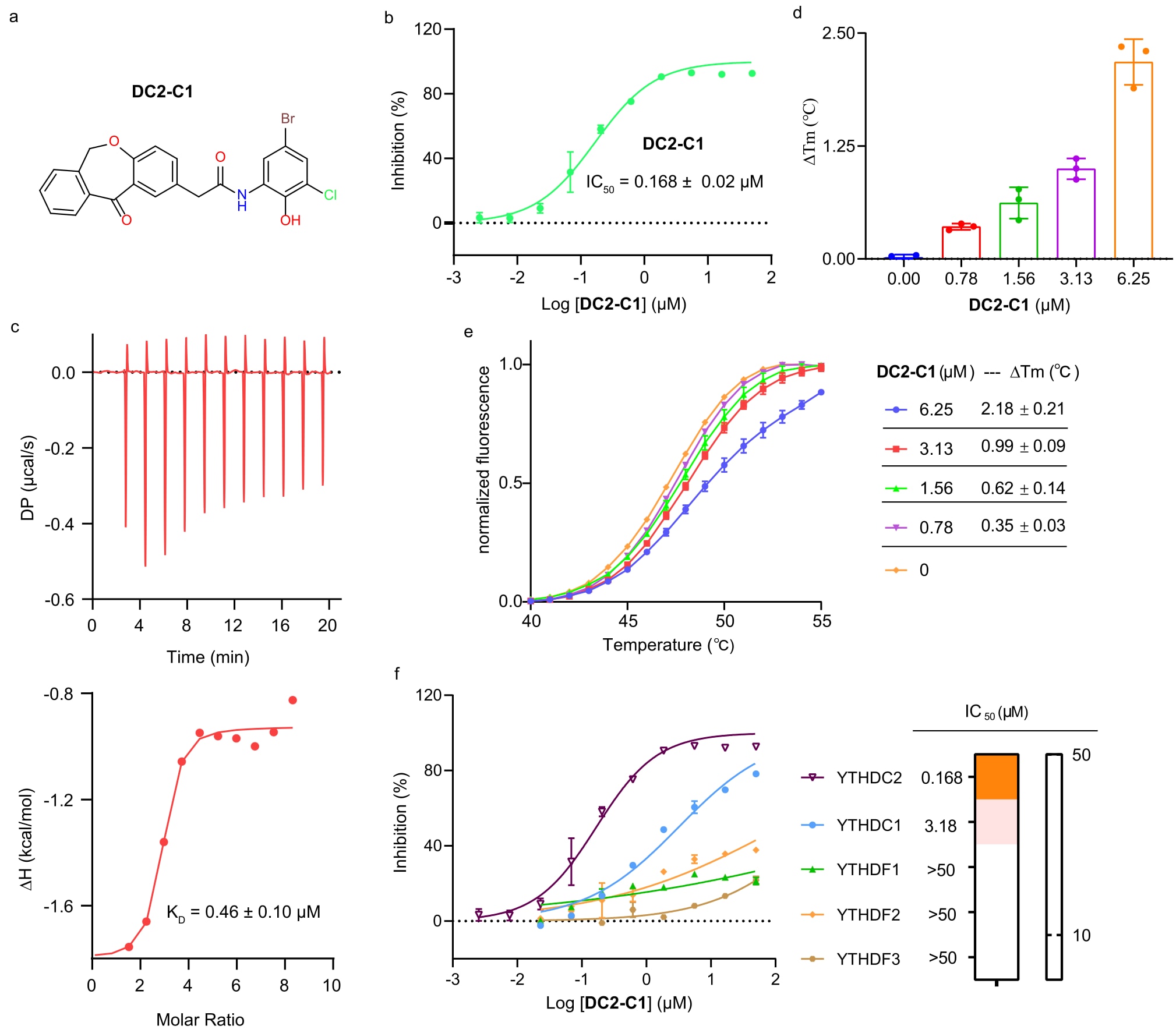
图5|DC2-C1 针对 YTHDC2 的生物活性 a. DC2-C1 的化学结构。 b. 基于 FP 实验测定的 DC2-C1 剂量-活性曲线。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 c. YTHDC2 与 DC2-C1 的 ITC 结合曲线。数据以三次独立实验的均值 ± 标准差表示。 d, e. 采用 DSF 检测 DC2-C1 对 YTHDC2 热稳定性的影响。DC2-C1 浓度从高到低依次为 6.25 µM、3.13 µM、1.56 µM、0.78 µM 和 0 µM。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 f. 基于 FP 实验测定的 DC2-C1 对 m6A“reader”蛋白的剂量-活性曲线。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。
为进一步验证DC2-C1的体外生物活性,采用多种生化与生物物理方法进行评估。等温滴定量热法测定其与YTHDC2的结合亲和力,结果显示其平衡解离常数KD为0.46 ± 0.10 μM,表明具有较强的结合能力。此外,差示扫描荧光实验表明,DC2-C1能够以剂量依赖方式稳定YTHDC2蛋白,在6.25 μM浓度下引起2.18 ± 0.21 °C的热稳定性提升。
进一步的荧光偏振筛选实验表明,DC2-C1对YTHDC2的抑制作用显著强于其他含YTH结构域的m6A读取蛋白,显示出良好的选择性。
2.7 DC2-C1与YTHDC2的结合模式
为探究DC2-C1与YTHDC2之间的可能结合模式,在共晶实验未成功的情况下,采用分子对接分析进行研究。由于命中化合物来源于m6A结合口袋,推测DC2-C1的结合位点与m6A结合位点重合。为获得更精确的结合构象,采用Glide进行诱导契合对接分析。结果显示,DC2-C1能够很好地嵌入YTHDC2的m6A结合口袋,从而可能阻断其与m6A-mRNA的结合。与YTHDC2-H3的对接结果相比,DC2-C1的结合模式与H3相似。
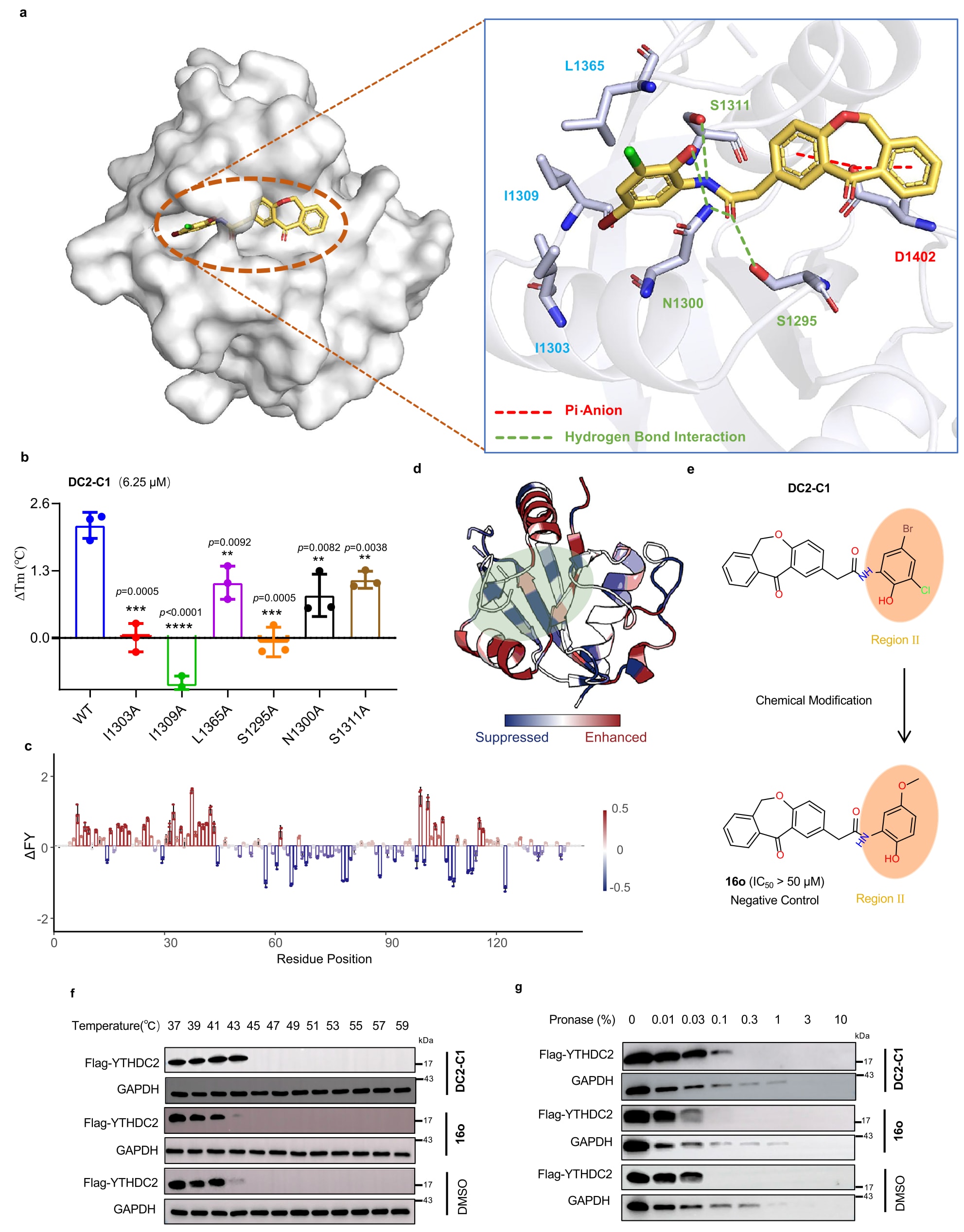
图6|DC2-C1 与 YTHDC2 的结合模式及其在活细胞中的靶标结合情况 a. DC2-C1 与 YTHDC2 的预测结合模式(YTHDC2 的三维结构来源于 PDB ID: 6K6U),蛋白质与分子结构由 PyMOL(Version: 3.1)绘制。 b. DC2-C1(6.25 µM)与重组突变型 YTHDC2 蛋白结合的 DSF 分析结果。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 c, d. 基于 a/x 离子总丰度,对 YTHDC2 7+ 与 YTHDC2-DC2-C1 7+ 的 UVPD 碎裂倾向进行比较,并映射至晶体结构。蓝色表示抑制,红色表示增强,绿色区域表示可能的结合口袋。数据以三次独立实验的均值 ± 标准差表示。 e. 针对 DC2-C1 的化学修饰示意图,提供阴性对照化合物 16o。 f. 在有或无 DC2-C1/16o 条件下,对转染 Flag-YTHDC2(YTH 结构域)的 HEK293T 细胞进行 CETSA 实验。所示条带为三次生物学重复的代表结果。 g. 在有或无 DC2-C1/16o 条件下,对转染 Flag-YTHDC2(YTH 结构域)的 HEK293T 细胞进行 DARTS 实验。所示条带为三次生物学重复的代表结果。(*P < 0.05,**P < 0.01,***P < 0.001,****P < 0.0001;采用双尾非配对 Student’s t 检验)。
具体相互作用分析表明,DC2-C1在m6A结合位点形成多种关键作用:其2-氨基-4-溴-6-氯苯酚中的羟基与N1300形成氢键;该结构同时与I1303、I1309及L1365发生疏水相互作用;酰胺键中的氮原子与S1311形成氢键,氧原子则与N1300和S1295形成氢键。此外,二苯并[b,e]氧杂卓结构中的两个苯环分别与D1402形成π-阴离子相互作用。
为验证该结合模式,进一步开展了突变实验。根据对接结果,选取D1402、I1303、I1309、L1365、N1300、S1311及S1295等关键残基进行突变分析。成功构建了I1303A、I1309A、L1365A、N1300A、S1311A及S1295A六种突变体,而D1402A由于蛋白稳定性问题未能获得。通过差示扫描荧光实验评估DC2-C1对这些突变体的作用,结果显示所有突变均显著降低其活性,从而支持上述结合模式的合理性。
进一步采用核磁共振技术分析结合位点。由于DC2-C1水溶性较差,难以获得有效数据,因此对其进行化学修饰,获得水溶性更好的类似物16p(IC50 = 1.88 ± 0.07 μM),并用于NMR分析。结果显示,N1297、N1300、L1301、K1307、T1312、T1313、K1319、A1323及I1352等氨基酸残基在结合过程中发生化学位移变化,这些残基位于或邻近m6A识别口袋,表明该区域为主要结合位点。
尽管上述结果表明类似物16p与m6A识别口袋结合,但由于其与DC2-C1结构存在差异,仍需进一步验证DC2-C1是否作用于同一位点。为此,采用原生自上而下质谱结合193 nm紫外光解离技术进行分析。该方法可在较低配体浓度下解析蛋白质结构变化及结合位点。在实验中,YTHDC2在接近天然状态下通过电喷雾电离进入质谱系统,随后对特定电荷态离子进行选择性分离并进行紫外光解离,从而获得结构信息。
结果显示,DC2-C1或16p结合后,YTHDC2大部分主链片段的解离效率降低,表明存在非共价相互作用。尤其是解离效率降低最显著的区域对应已知的m6A结合口袋。这一结果进一步证明,DC2-C1及其类似物均结合于YTHDC2的m6A识别位点。
2.8 DC2-C1在活细胞中与YTHDC2的结合
进一步对DC2-C1的细胞水平活性进行评估。在实验前,合成了一个阴性对照化合物16o,其为DC2-C1的结构类似物,通过去除卤素原子并引入甲氧基获得。在荧光偏振实验中,16o对YTHDC2未表现出明显抑制活性(IC50 > 50 μM),且未观察到荧光干扰。根据预测的结合模式推测,其无活性可能源于甲氧基与YTHDC2中I1309残基之间的空间位阻冲突。
采用细胞热转移实验(CETSA)与药物亲和响应靶点稳定性实验(DARTS)评估DC2-C1在活细胞中与YTHDC2的直接相互作用。由于YTH结构域仅占YTHDC2蛋白的一小部分,直接检测全长蛋白可能无法准确反映该结构域的结合情况。因此,在实验中使用转染pCDH-GFP-3xFlag-YTHDC2(YTH结构域)的HEK293T细胞。结果显示,DC2-C1处理显著提高了YTHDC2蛋白的热稳定性,并能够有效抑制蛋白酶诱导的降解,表明其与YTHDC2存在直接结合。相比之下,16o在上述实验中未表现出任何作用。
需要指出的是,在CETSA与DARTS实验中采用过表达YTH结构域的细胞模型存在一定局限,可能无法完全反映DC2-C1在野生型细胞中的真实结合情况。因此,后续研究将进一步在野生型细胞体系中验证其对YTHDC2的调控作用。
2.9 DC2-C1在RA-FLS中的体外细胞活性
如前所述,YTHDC2通过促进类风湿性关节炎成纤维样滑膜细胞(RA-FLS)的异常增殖与侵袭行为,加速关节破坏,从而推动类风湿性关节炎的进展。抑制YTHDC2的m6A结合能力会降低其靶基因AMIGO2 mRNA的稳定性,进而减少AMIGO2蛋白表达,从而抑制RA-FLS的增殖、迁移与侵袭行为,缓解疾病进展。与此一致,通过CRISPR技术敲除MH7A细胞(人源RA-FLS细胞系)中的YTHDC2,可显著降低AMIGO2蛋白水平。
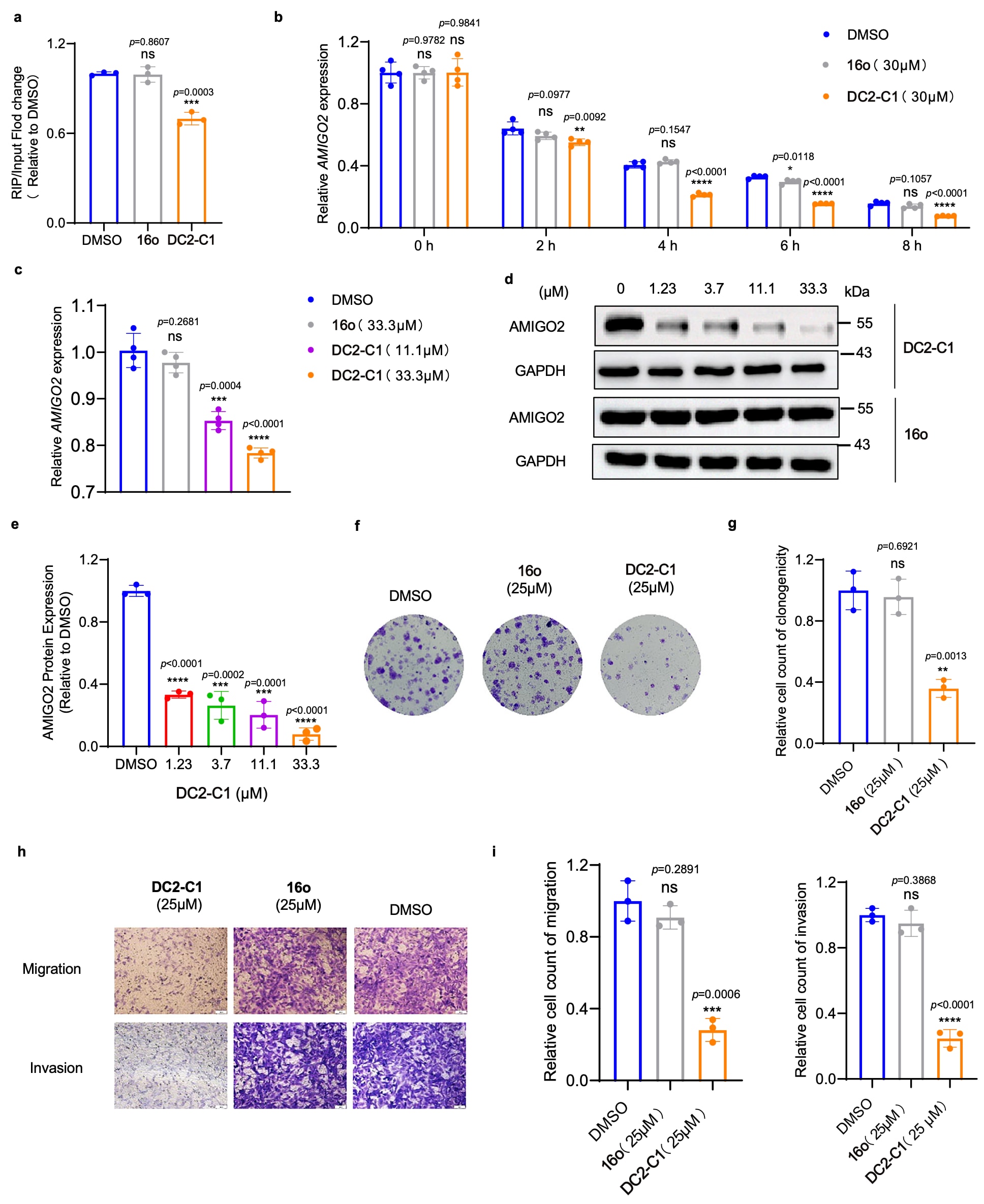
图7|DC2-C1 在 RA-FLS 中的生物活性评估 a. 通过 RIP-qPCR 实验分析 YTHDC2 与 AMIGO2 mRNA 在 RA-FLS 中的相互作用(处理条件为 DMSO、DC2-C1 或 16o)。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 b. 在加入放线菌素 D 后,于不同时间点检测经 DC2-C1 或 16o 处理的 RA-FLS 中 RNA 降解情况(以 0 h 归一化)。数据以四次独立实验中每次四次技术重复的均值 ± 标准差表示。 c. 通过 RT-qPCR 验证经 DC2-C1 或 16o 处理的 RA-FLS 中 AMIGO2 mRNA 表达水平。数据以四次独立实验中每次四次技术重复的均值 ± 标准差表示。 d. 通过 Western blot 分析经 DC2-C1 或 16o 处理的 RA-FLS 中 AMIGO2 蛋白表达。所示条带为三次生物学重复的代表结果。 e. 对 MH7A 细胞 Western blot 结果进行定量分析。数据以三次独立实验的均值 ± 标准差表示。 f. 经 DC2-C1 或 16o 处理的 MH7A 细胞集落形成实验。 g. 对 MH7A 细胞集落形成进行定量分析。数据以三次独立实验的均值 ± 标准差表示。 h. 经 DC2-C1 或 16o 处理的 MH7A 细胞迁移与侵袭能力。比例尺,50 μm。 i. 对 MH7A 细胞迁移与侵袭进行定量分析。数据以三次独立实验的均值 ± 标准差表示。(*P < 0.05,**P < 0.01,***P < 0.001,****P < 0.0001;采用双尾非配对 Student’s t 检验)。
进一步通过RIP-qPCR实验评估DC2-C1对YTHDC2与AMIGO2 mRNA结合的影响。结果显示,DC2-C1处理显著降低了MH7A细胞中YTHDC2与AMIGO2 mRNA的结合能力。RNA稳定性实验表明,DC2-C1显著加速了AMIGO2 mRNA的降解,并伴随AMIGO2 mRNA水平的剂量依赖性下降以及蛋白表达的减少,而GAPDH表达未受影响。相比之下,阴性对照化合物16o在上述实验中未表现出明显作用。这些结果表明,DC2-C1在细胞内抑制了YTHDC2与AMIGO2 mRNA的结合,削弱其稳定作用,从而降低AMIGO2表达。但不能完全排除DC2-C1通过其他调控机制影响AMIGO2表达的可能性。
最后,通过功能实验评估DC2-C1对MH7A细胞表型的影响。在克隆形成实验中,DC2-C1显著抑制了细胞克隆形成能力;Transwell实验结果显示,其明显降低了细胞的迁移与侵袭能力。在所有实验中,阴性对照化合物16o均未表现出显著影响。
2.10 DC2-C1在PDAC细胞与肝细胞中的体外细胞活性
在胰腺导管腺癌(PDAC)细胞中进一步评估DC2-C1的作用。已有研究表明,敲除YTHDC2可抑制该类细胞的恶性表型,其机制在于YTHDC2通过m6A依赖方式促进组蛋白H3赖氨酸4三甲基化(H3K4me3),从而增强染色质可及性并促进致癌基因表达。与此一致,在PDAC细胞中通过CRISPR敲除YTHDC2可显著降低H3K4me3水平。对PANC-1与MiaPACA-2细胞进行DC2-C1处理后,观察到H3K4me3表达呈剂量依赖性下降。在功能层面,DC2-C1显著抑制了两种细胞系的克隆形成能力与侵袭能力,而阴性对照化合物16o未表现出明显抑制作用。
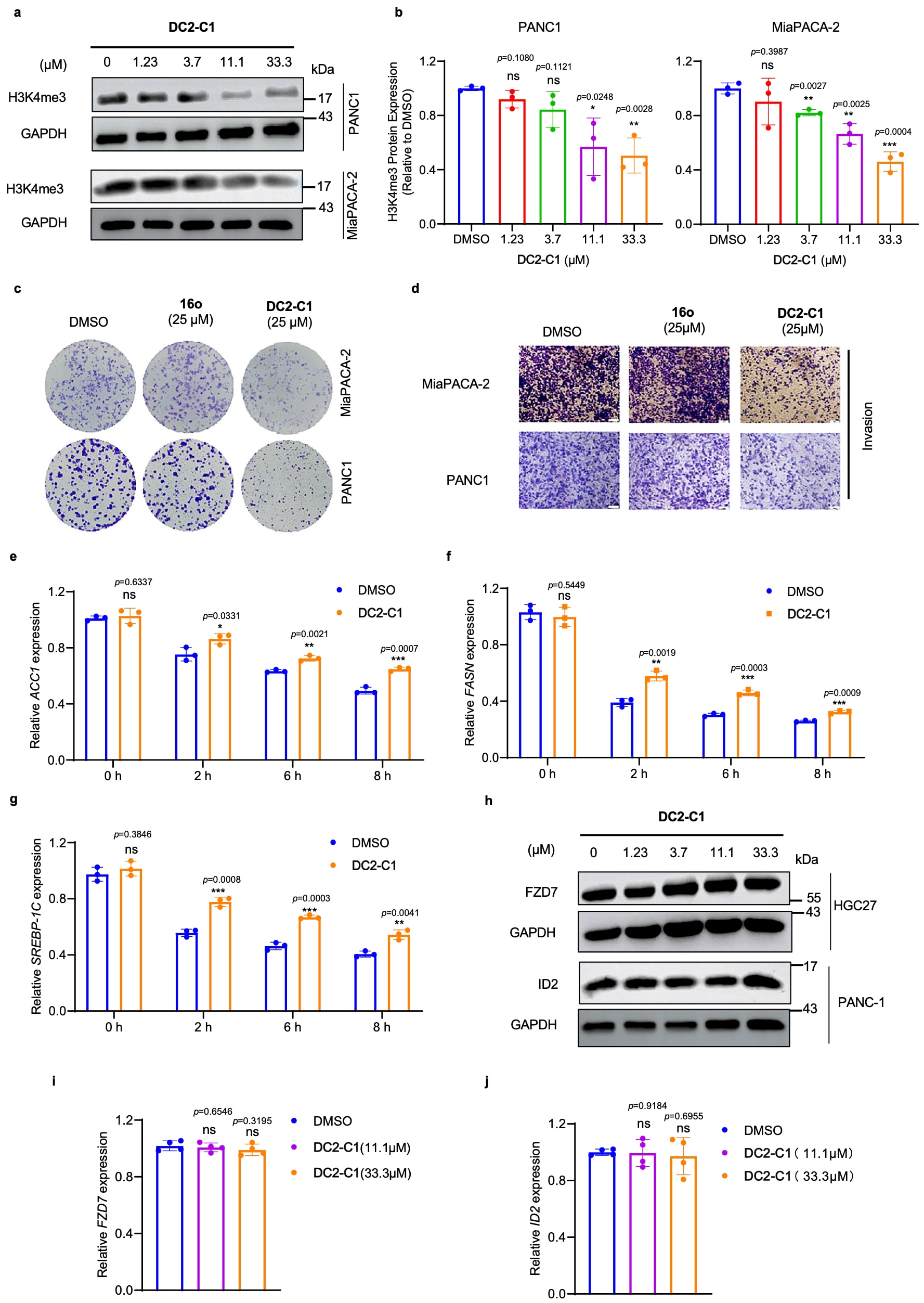
图8|DC2-C1 在 PDAC 细胞与肝细胞中的体外生物活性 a. DC2-C1 处理的 PANC-1 或 MiaPACA-2 细胞中 H3K4me3 表达的 Western blot 分析。所示条带为三次生物学重复的代表结果。 b. 对 PANC-1 或 MiaPACA-2 细胞 Western blot 结果进行定量分析。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 c. 经 DC2-C1 或 16o 处理的 PANC-1 或 MiaPACA-2 细胞集落形成实验。 d. 经 DC2-C1 或 16o 处理的 PANC-1 或 MiaPACA-2 细胞侵袭能力。比例尺,50 μm。 e–g. 在 DC2-C1 处理的 HepG2 细胞中,通过 RT-qPCR 测定 ACC1、FASN 和 SREBP-1C 的 mRNA 表达。数据以三次独立实验中每次三次技术重复的均值 ± 标准差表示。 h. DC2-C1 处理的 PANC-1 细胞中 ID2 表达的 Western blot 分析;DC2-C1 处理的 HGC27 细胞中 FZD7 表达的 Western blot 分析。所示条带为三次生物学重复的代表结果。 i. 在 DC2-C1 处理的 HGC27 细胞中,通过 RT-qPCR 测定 FZD7 mRNA 表达。数据以四次独立实验中每次四次技术重复的均值 ± 标准差表示。 j. 在 DC2-C1 处理的 PANC-1 细胞中,通过 RT-qPCR 验证 ID2 mRNA 表达。数据以四次独立实验中每次四次技术重复的均值 ± 标准差表示。(*P < 0.05,**P < 0.01,***P < 0.001,****P < 0.0001;采用双尾非配对 Student's t 检验)。
在肝细胞模型中,YTHDC2在调控脂质生成及甘油三酯稳态方面具有重要作用。已有研究显示,在HepG2细胞中敲低YTHDC2会导致关键脂质生成相关基因ACC1、FASN与SREBP-1C的mRNA水平上调。通过CRISPR敲除YTHDC2验证了上述结果。相应地,DC2-C1处理同样引起ACC1、FASN与SREBP-1C mRNA水平的剂量依赖性升高。
最后,对DC2-C1在活细胞中的选择性进行了评估。选取两个并非YTHDC2靶标、但分别受其他YTH蛋白调控的mRNA作为对照,即YTHDF1靶标FZD7与YTHDF2靶标ID2。实验结果显示,DC2-C1处理未改变HGC27细胞中FZD7以及PANC-1细胞中ID2的mRNA与蛋白表达水平,表明其在细胞水平具有良好的选择性。
3 讨论
RNA的m6A修饰在多种生理与病理过程中发挥关键作用。调控m6A修饰的相关蛋白失调已被证实与多种疾病密切相关,包括免疫疾病、肿瘤、代谢综合征及神经系统疾病。因此,开发针对m6A调控蛋白的抑制剂,对于相关重大疾病的治疗及机制研究具有重要意义。目前,已有多种针对甲基转移酶METTL3、去甲基化酶FTO与ALKBH5,以及其他YTH家族蛋白(YTHDF1-3与YTHDC1)的高效抑制剂被报道,并在多种疾病中展现出良好的治疗潜力。然而,针对YTHDC2的高效抑制剂仍未见报道。
针对YTHDC2这类新靶点筛选活性化合物具有较大挑战。为在避免高通量筛选高成本的前提下快速获得命中化合物,引入基于人工智能的分子生成模型进行配体设计。为此构建了EPMolGen模型,该模型通过显式引入静电特征区别于现有方法。消融实验结果表明,引入静电信息能够显著提升模型性能。利用该模型生成针对YTHDC2的小分子抑制剂,从中筛选并合成了5个候选化合物,其中1个表现出IC50为16.84 μM的活性。通过进一步的结构优化与构效关系分析,获得高效抑制剂DC2-C1,其IC50达到0.168 μM。该化合物对其他YTH蛋白具有良好选择性,且细胞毒性较低。同时,DC2-C1可显著降低多个YTHDC2靶基因的表达,从而抑制相关细胞表型。
需要指出的是,DC2-C1目前仍属于先导化合物,仍需进一步优化。体外药代动力学与毒性预测结果表明,其具备一定类药性,但仍存在水溶性较差及可能抑制部分代谢酶等问题。此外,当前版本的EPMolGen仅考虑了受体蛋白的静电特征,这一设计在自回归流模型框架下已显著提升性能。未来可进一步引入配体分子的静电特征,以实现对相互作用的更全面建模,从而进一步提升生成效果。
总体而言,该分子生成模型成功实现了针对YTHDC2的高效且具有细胞活性的先导化合物发现,展示了深度学习在药物发现早期阶段中的应用潜力。