Nat. Protoc. 2025 | 用于快速且精确比较蛋白质和核酸结构的US-align图形界面与命令行协议
今天介绍的这项工作来自 Nature Protocols 。该研究围绕结构生物信息学中一个核心问题展开,即如何在蛋白质与核酸等生物大分子之间实现快速且准确的三维结构比较。随着深度学习结构预测方法的快速发展,大量高质量结构模型不断涌现,使得结构比对从单一任务转变为高通量、跨分子类型的基础分析工具。在这一背景下,传统方法因针对特定分子类型或特定任务设计,逐渐难以满足统一分析与规模化应用的需求。该文提出的US-align方法通过引入统一的评分函数TM-score,并结合迭代结构叠合与动态规划策略,将蛋白质、RNA和DNA的多种结构比对任务整合到同一框架中。该方法不仅支持单体、复合物以及多结构比对,还能够处理非顺序对齐等复杂情形,从而实现跨分子类型的结构比较。这种统一性对于功能注释、模板建模以及分子模拟研究具有重要意义。在性能方面,US-align在保持高精度的同时显著提升了计算效率,能够在极短时间内完成结构比对,适用于大规模数据分析。同时,方法提供了命令行工具、在线服务以及与分子可视化软件的集成接口,使其在不同使用场景下均具有良好的可操作性。总体来看,该工作不仅提出了一种高效实用的结构比对工具,也为不同类型生物大分子之间的统一分析提供了方法学基础,对结构生物学与计算生物学研究具有重要参考价值。

获取详情及资源:
0 摘要
随着结构生物学的发展以及基于深度学习的结构预测技术不断进步,大分子结构之间的快速且准确比较在结构生物信息学中变得愈发重要。US-align是一种高效、通用且开源的程序,可用于蛋白质、RNA和DNA的顺序与非顺序结构比对,支持成对比对和多结构比对,并适用于单体以及多聚复合物结构。US-align的核心算法建立在高度优化的迭代结构叠合与动态规划比对过程之上,并由统一且与序列长度无关的评分函数
1 引言
1.1 协议的发展
分子层面的结构比较,包括结构叠合与结构比对,是结构生物学和生物信息学中的基础问题。其中,结构叠合通常是在已知序列对应关系(即残基级对应关系)的前提下,通过优化
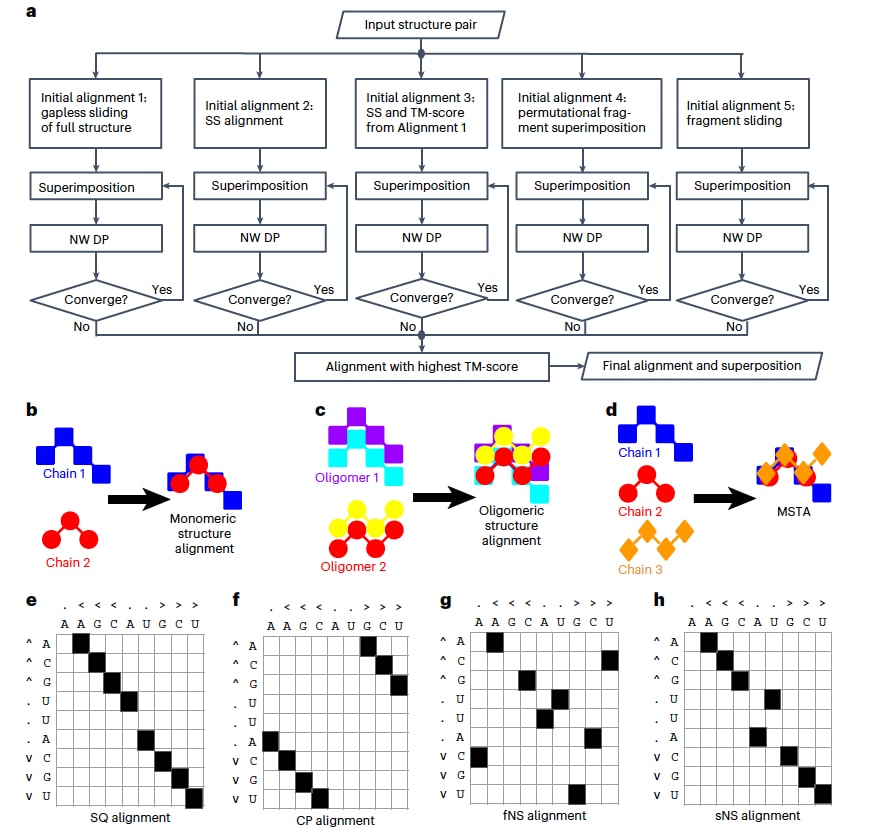
图1|US-align算法及其不同对齐模式示意图。 a,US-align用于不同对齐任务的核心迭代“结构叠合-比对”算法。其中,“结构叠合”指通过对一个结构进行旋转和平移,使结构对之间的
1.2 方法的应用
US-align已在学术界被广泛用于大分子结构比较,例如在多种结构预测竞赛中用于模型质量评估。其应用并不仅限于结构相似性分析,准确的结构比对还可服务于多种研究任务,包括基于结构的功能注释、蛋白质-配体相互作用预测、蛋白质-蛋白质及蛋白质-RNA对接、蛋白结构域组装、基于进化的蛋白设计、片段引导的结构模型优化以及生物信息学数据库的构建与整理等。
1.3 与其他方法的比较
尽管US-align并非针对某一特定结构比对任务的唯一方法,也不是唯一基于
虽然部分数据库检索程序在速度上更快,但其性能依赖于预先构建和筛选的结构数据库,并需要对目标结构进行预计算表示,因此并不适用于未知结构之间的直接比较,而US-align则可直接用于通用结构比对任务。
1.4 实现该协议所需的基础
US-align提供三种主要使用方式,包括命令行工具、在线Web服务器以及分子图形系统插件(如PyMOL)。其中,Web服务器和PyMOL插件无需命令行操作,使用方式直观,适合一般结构生物学用户。而在本地计算机上安装并运行命令行版本,则需要具备一定的操作系统环境下的命令行基础知识。
1.5 局限性
尽管US-align具有较强的通用性,但其并不适用于柔性结构比对。在某些情况下,例如同源结构之间存在较大构象变化(如结构域取向变化)时,需要使用专门的柔性比对工具,如FATCAT或Matt。此外,虽然US-align提供了PyMOL插件,但目前尚未支持如UCSF ChimeraX等先进分子可视化软件的插件接口,这些功能有待未来进一步扩展和完善。
1.6 实验设计
为展示US-align在不同场景下的应用,选取了若干具有代表性的结构对作为示例。在单体结构比对中,首先选取两种肌红蛋白(PDB ID: 101m与1mba),其序列相似性较低(约24.5%),但结构相似性较高(
在多聚体结构比对中,选取了一对较难对齐的八聚体结构(PDB ID: 4jhm与4iaj),其序列相似性极低(约5.3%),结构相似性中等(
这些示例主要用于展示复杂比对情形及结构可视化效果。实际应用中,只要提供PDB或mmCIF格式的三维结构文件,均可使用US-align完成相应的结构比对任务。